| Titel: | Ueber die quantitative Bestimmung des Gesammtkohlenstoffs im Gußeisen und Stahl; von Dr. Julius Löwe in Frankfurt a. M. |
| Autor: | Julius Löwe [GND] |
| Fundstelle: | Band 148, Jahrgang 1858, Nr. C., S. 432 |
| Download: | XML |
C.
Ueber die quantitative Bestimmung des
Gesammtkohlenstoffs im Gußeisen und Stahl; von Dr. Julius Löwe in Frankfurt a. M.
Mit einer Abbildung auf Tab. VII.
Lowe, über die quantitative Bestimmung des Gesammtkohlenstoffs im
Gußeisen und Stahl.
Gedenkt man des bekannten Verhaltens des festen metallischen Jods zu metallischem
Eisen, der Eigenschaft des ersteren nämlich: ohne Zersetzung von Wasser und somit
ohne Entbindung von Wasserstoffgas sich mit letzterem direct zu Eisenjodür oder bei
Ueberschuß von Jod zu Eisenjodid zu verbinden, so liegt nach Betracht dieses in der
That der Gedanke nahe: daß man hierauf eine Methode zur Bestimmung der Gesammtmenge
des Kohlenstoffs im Gußeisen oder Stahl gründen kann, insofern ja der gebundene wie
der in Form von Graphit gelöste Kohlenstoff des Eisens beide unberührt vom Jod mit
ihrer charakteristischen Eigenschaft neben der gebildeten Lösung des Eisenjodürs
zurückbleiben. Diese schon von Berzelius in seinem
Lehrbuch der Chemie Bd. III S. 458 erwähnte Methode wurde von diesem in so weit
nicht für zweckmäßig erachtet, als die Einwirkung des Jods auf das metallische Eisen
zu langsam und träge erfolge. Allein dieser gemachte
Einwurf ist nur dann gerechtfertigt wie gegründet, wenn man die mit Wasser
befeuchtete Eisenprobe zur Bildung der Verbindung dem festen Jod in stärkeren, soliden Stückchen
darbietet, hebt sich jedoch auf, sobald man bei der Ausführung nicht außer Acht
läßt, das Metall in Form von sehr feinen Feilspänen den Angriffen des Jods
preiszugeben. Nach mehreren Versuchen fand ich die Einwirkung des Metalloids unter
dieser Berücksichtigung dann so rasch und energisch, daß zur völligen Auflösung des
Eisenmetalls nicht mehr Zeit, als bei den übrigen bis jetzt bekannten Methoden
erforderlich war, ja, deren Betrag sogar als noch kürzer gefaßt werden kann, indem
z.B. 5 Gran in Anwendung gebrachter, fein zertheilter Stahl kaum mehr als eine
Stunde zur völligen Auflösung des Eisens und Ausscheidung des enthaltenen
Kohlenstoffs bedurften. Allerdings kann hier bei Befolgung dieser Methode nicht in
Abrede gestellt werden, daß die Sättigungscapacität des Jods im Vergleiche zum Eisen
sehr gering ist – ein Einwand, welcher gleichfalls von Berzelius mit mehr Recht gemacht wurde, denn zur Bildung von Eisenjodür
von der Zusammensetzung = FeJ verlangt 1 Aeq.
metallisches Eisen, repräsentirt durch die Zahl 28, eine gleiche Repräsentation von
1 Aeq. festem metallischem Jod, ausgedrückt durch die Zahl = 127, oder die
reducirten Gewichtsverhältnisse beider stehen, kürzer gefaßt, unter sich beim
Eintritte der Verbindung nahe in der Proportion 1 : 4,6, so daß also auf je 1
Gewichtstheil metallisches Eisen oder hier Stahl oder Gußeisen als solches
angesehen, 4,6 Gewichtstheile metallisches Jod zum Zwecke völliger Verbindung und
Lösung in Anwendung zu bringen sind. Da sich das bei dieser Methode verbrauchte Jod
in Gestalt von Jodkalium leicht wieder gewinnen läßt, ein Salz, in dessen Form das
Jod in den Laboratorien die häufigste Anwendung erleidet, so dürfte, die
Kostspieligkeit etwa hier in Anschlag gebracht, auch in dieser Richtung der
praktischen Ausführung genannter Methode kein erhebliches Hinderniß im Wege stehen.
Ferner scheint das metallische Jod nicht ohne weitere Einwirkung auf das vorhandene
Schwefelarsen- und Phosphor-Eisen zu seyn, Bestandtheile, welche dem
Gußeisen und Stahl als Begleiter so häufig in geringer Menge beigesellt sind.
Wahrscheinlich erzeugen sich bei diesem Processe Verbindungen des Jods mit Phosphor
und Arsen, vielleicht Arsensuperjodür und phosphorige Säure oder Phosphorsäure unter
Entbindung geringer Mengen von Jodwasserstoffgas, indem die Verbindungen des Jods
mit Phosphor durch Wasser bekanntlich in genannte Producte zerlegt werden. Wenn
schon diese hier ausgesprochene Behauptung durch genauere Data zu rechtfertigen ist,
als hier geschehen, wo ich mir einstweilen nur vermuthungsweise erlaubt habe diese
Ansicht auszusprechen, bis genauere Versuche mit Phosphoreisen, Arseneisen etc. und
deren Verhalten zu freiem Jod mich mehr zu einer solchen Annahme berechtigen, so haben mich
doch directe Versuche mit geschmolzenem Schwefeleisen, wie solches in dem
Laboratorium zur Entwickelung von Schwefelwasserstoffgas dient, zu dem Resultate
geführt, daß dieses letztere wenigstens vollständig durch festes Jod bei Gegenwart
von Wasser unter Wärmeentwickelung und Ausscheidung von gelblich gefärbtem Schwefel
zerlegt wird. Dann als ich fein gepulvertes Schwefeleisen von oben genannter Art
mittelst starker Salpetersäure oxydirte und die so gebildete Schwefelsäure mittelst
salpetersaurem Baryt aus der Lösung nach bekannten Regeln ausfällte, wurde
erhalten:
Schwefelsaurer Baryt
=
1,5884
Schwefelsäure =
0,5457
Schwefel =
0,2183
Procente Schwefel =
20,517.
Nachdem ich nun eine gleiche Probe erwähnten Schwefelmetalls mit Wasser und
metallischem Jod behandelte und nach dem völligen Auswaschen und Entfernen des
Eisenjodürs und überschüssigen Jods den Rückstand mit Salpetersäure oxydirte, wurde
die Menge des Schwefels auf gleiche Art, wie oben bestimmt, aus dem erhaltenen
schwefelsauren Baryt zu 20,496 Proc. berechnet, eine Zahl welche in völliger
Uebereinstimmung mit der zuerst gefundenen Gewichtsmenge des Schwefels steht. Eine
Verbindung von Jodschwefel scheint also hier bei Behandlung auf kaltem Wege nicht
erzeugt zu werden, wofür auch die lichtgelbe Farbe des Rückstandes nach der
Einwirkung von Jod spricht, indem alle bis jetzt bekannten Producte des Jods mit
Schwefel in verschiedenen Mengenverhältnissen ein mehr schwarzgraues Ansehen
besitzen. Auch gab eine auf ähnliche Art erhaltene Probe nach dem Trocknen beim
Erhitzen nur gelbliche beim Erkalten erstarrende Tröpfchen von reinem sublimirten
Schwefel ohne violette Dämpfe von Jod. Läßt man hingegen das metallische Jod in der
Wärme auf das Schwefeleisen einwirken, oder trägt man zu etwas größeren Mengen des
letzteren den ganzen zur Lösung des Eisens und zur Bildung von Eisenjodür
nothwendigen Antheil von Jod auf einmal hinzu, wobei die Verbindung ebenfalls unter
Wärmeentwickelung erfolgt, so besitzt oft der resultirende Rückstand ein mehr graues
Ansehen und dann einen Gehalt von Jod. Es ist nach diesen Thatsachen nicht gerade
unwahrscheinlich, daß auch eine Zerlegung des Phosphoreisens, Arseneisens u.s.w.
durch Jod zu bewerkstelligen ist, indem z.B. auch andere in Salzsäure fast
unlösliche Phosphormetalle, wie unter anderen das Phosphorkupfer, durch Jod unter
Bildung von Jodwasserstoffsäure und Phosphorsäure zersetzt werden, welche
interessante Thatsache bekanntlich Böttger bei
Beschreibung der Darstellung von Phosphortupfer aus Phosphor und einer wässerigen
Lösung von Kupfervitriol in der Siebhitze mitgetheilt hat.Jahresbericht des physikalischen Vereins zu Frankfurt a. M. für
1855–1856, S. 32; polytechn. Journal Bd. CXLIV. 203.
Bei Ausführung der Analyse verfährt man nun auf folgende Art: Das zur quantitativen
Untersuchung auf den Gesammtkohlenstoff in größeren Stücken vorliegende Gußeisen
oder der Stahl wird mittelst einer scharfen Feile in feine Feilspäne geraspelt,
welch letztere man durch dichte Leinwand beutelt, um so mechanisch die feineren
Theilchen des Metalls von den gröberen abzusondern. Aus diesem schon fast zarten
Pulver zieht man mittelst eines kleinen Magneten die metallischen Eisenpartikel
heraus, um dieselben von kleineren anhängenden Verunreinigungen, wie etwa in der
Masse mechanisch eingeschlossen gewesenen kleinen Quarzkörnchen oder durch die
Operation des Feilens etwa zugeführten geringen Mengen von Oxyd u. dgl. auf diese
Weise sorgfältig zu trennen.
Zur Bestimmung des Kohlenstoffgehaltes verschiedener so zertheilter Stahlproben
wurden für je eine Analyse 5–6 Grm. in Anwendung gebracht. Man gibt jedes für
sich abgewogene Quantum des Metalls in ein geräumiges mehr hohes als weites
Becherglas, übergießt es darauf mit 200 Kub. Centim. ausgekochten Wassers und trägt
nachher das der genannten angewandten als reines Eisen angenommenen Menge
entsprechende Gewichtsquantum von reinem metallischem Jod im Betrage von
23–27,6 Grm. in die Flüssigkeit ein. Gibt man sogleich die ganze Menge des
Jods hinzu, so geht die Vereinigung zwischen Jod und Eisen unter Entbindung von
Wärme etwas zu lebhaft vor sich, wodurch Gefahr vorhanden wäre, daß Theilchen aus
dem zur Auflösung dienenden Gefäße herausgeschleudert werden und somit ein Verlust
zu befürchten stünde. Aus diesem Grunde ist die successive Zugabe des Jods und die
Sorge für Abkühlung des Becherglases dem plötzlichen Eintragen vorzuziehen. Sobald
man mittelst des Glasstabes keine rauhen, knirschenden Körnchen am Boden des Glases
nach Ablauf etwa einer Stunde mehr wahrnimmt, so ist die Auflösung und Bildung der
Verbindung beendet, wozu in den seltensten Fällen ein größerer Aufwand von Zeit
erforderlich ist. Besitzt nach diesem Intervall die Flüssigkeit eine tief braune
Farbe, so ist man sicher die nöthige Menge von Jod in Anwendung gebracht zu haben,
was wohl immer der Fall seyn dürfte, indem man ja bei der Berechnung der zur
Vereinigung nöthigen Gewichtsverbindungen von Jod und Eisen von der Annahme eines
reinen angewandten Metalls ausging. Besäße dessen ungeachtet die Lösung nur eine
grünliche Farbe von Eisenjodür, so dürfte selbstverständlich der Zusatz von Jod in
geringer Menge erneuert werden, so lange zwar, bis obiger Bedingung Genüge
geschehen.
Wir gehen hier freilich von der Voraussetzung aus: daß das mit dem Eisen in
Wechselwirkung tretende Jod vollständig rein und also frei von einem Gehalte
wenigstens organischer Substanzen ist, nicht etwa gemengt mit kleinen
Holzpartikelchen und dergleichen wie Jod des Handels auf ähnliche Art verunreinigt
mir schon öfters in die Hände gekommen. Beim Eintreten des letzteren Falls löst man
am besten das metallische Jod in einer mäßig concentrirten Lösung von Jodkalium auf,
und filtrirt dieselbe direct in das die abgewogene Eisenprobe enthaltende
Becherglas. Ist der Proceß der Auflösung auf diese Art beendet, so filtrirt man die
braune jodhaltige Flüssigkeit durch ein anfangs mit verdünnter Salzsäure und Wasser
gewaschenes, darauf bei 112° C. im Luftbade getrocknetes und zwischen
verschlossenen Uhrgläsern gewogenes Filter mit der Vorsicht, daß der fast schwarze
durch die Einwirkung von Jod ungelöst gebliebene Schlamm von fein zertheilter Kohle
nicht aufgerührt wird, wascht den Rückstand und das Filter mit einer wässerigen
Lösung von Jodkalium mehrmals ab, kocht ihn darauf mit Wasser und schwefligsaurem
Natron zur Entfernung von etwas freiem ausgeschiedenem Schwefel bei einem Gehalte
der Probe von Schwefeleisen aus, darauf behandelt man ihn mit etwas Aetzammoniak und
zuletzt noch mit einer mäßig starken Lösung von reinem kohlensaurem Kali, und
endlich längere Zeit mit heißem destillirtem Wasser. Nach diesen aufeinander
folgenden Operationen sammelt man den so gereinigten zarten Schlamm auf oben
genanntem Filter von bekanntem Gewichte, trocknet ihn im Luftbade bei 110° C.
und wiegt, wie anfangs geschehen, das Filtrum mit seinem trockenen Inhalte nun
zwischen den mittelst einer Messingklemme verschlossenen Uhrgläsern. Der durch die
zweite Wägung gefundene Gewichtsüberschuß ergibt den wahren Gesammtgehalt an Kohle.
Man verbrennt darauf das Filtrum von bekanntem Aschegehalt nebst Inhalt in einem
gewogenen Platintiegel und ermittelt durch Wägung den Betrag der rückständigen
Asche. Ergibt sich nach Abzug dabei ein höherer Werth, als der bekannte des Filters,
so muß man diesen von dem Gewichte der anfänglich gefundenen Kohle abziehen, bevor
man deren Ertrag in Procente auswirft. Allerdings kann diese Ermittelung des
Kohlenstoffgehaltes nur einen Anspruch auf ein etwa annäherndes Resultat geben und
die dadurch gefundene Kohle vielleicht als roher Gesammtkohlenstoff, wie ich ihn
nannte, bezeichnet werden, denn es ergeben sich Differenzen bei Stahl zwischen dem
wahren nach vorgenommenen Prüfungen von 0,2–0,3 Proc. Es steht sogar zu
erwarten, daß diese
Unterschiede sich bei einzelnen Eisensorten noch beträchtlich höher stellen, je nach
dem Gehalte an Silicium, welches dem Gußeisen oder Stahle beigemengt ist, denn
dieses scheint sich indifferent gegen die Einwirkung des metallischen Jods zu
verhalten, geht somit wahrscheinlich nicht in die Lösung über, sondern bleibt der
gleichfalls unlöslichen Kohle beigemischt. Wenn der Werth dieses zuletzt genannten
Elementes in der That die Höhe von 1, ja sogar von 1,6 Proc. erreichen kann, wie
dieses neuerdings von Max Büchner und auch Anderen in
einer Probe schaumigen grobkörnigen Roheisens von Blansko gefunden wurde,Journal für praktische Chemie Bd. LXXII. S. 366; polytechn. Journal Bd. CXLVII. S. 288. so ist allerdings nach solchen Resultaten zu behaupten, daß die directe
Gewichtsermittelung der aus dem Gußeisen oder Stahl abgeschiedenen Kohle unstatthaft
und ungenau ist. Außerdem schließt diese Voraussetzung und Bestimmung ja noch eine
neue Fehlerquelle in sich, selbst wenn der Siliciumgehalt als = 0 angenommen werden
könnte, indem die abgeschiedene Kohle nicht als elementäre Kohle anzusehen ist,
sondern stets ein veränderlicher Bruchtheil vom Gesammtgewichte der letzteren dem
gebundenen Wasserstoffe zugehört. Nach diesen Erfahrungen steht es somit fest, daß
der wahre Werth des Kohlenstoffgehaltes nur durch Verbrennung und Oxydirung der
abgeschiedenen Kohle, Auffangen und Wiegen der gebildeten Kohlensäure und endlich
durch Zurückberechnung aus dieser letzteren Verbindung sich in Wahrheit ermitteln
läßt. In der Art der Ausführung war man daher bis jetzt immer genöthigt auf die
Methode der organischen Elementaranalyse zurückzukommen. So genau in der That deren
Ergebnisse nach vorliegenden Bestimmungen gefunden wurden, so läßt sich wiederum auf
der anderen Seite nicht in Abrede stellen, daß diese zuletzt genannte Methode etwas
zu mühsam und zeitraubend ist, um in dieser Beziehung mit gewünschtem Erfolge in den
häufigsten Fällen für die Praxis Anwendung zu finden. Die Beobachtung der Gebrüder
Rogers, nach welcher die Kohle selbst in ihrer
dichtesten Modification sich leicht durch eine Mischung von Schwefelsäure und saurem
chromsaurem Kali in der Wärme zu Kohlensäure oxydiren lasse, gibt in der That ein
Mittel an die Hand diese Bestimmung, ohne der Genauigkeit den geringsten Eintrag zu
thun, mit dem gewünschten Erfolge in Ausführung zu bringen, und kann daher
unbestritten zu derartigen Untersuchungen als Basis dienen. Wenn schon diese
Thatsache der völligen Umwandlung und Oxydirung des Kohlenstoffes nach dieser
Methode feststeht, so
ist ein nicht minder wichtiger Factor bei der praktischen Anwendung im Auge zu
behalten, der nämlich: einen Apparat in der Weise zu construiren, daß er allen
gestellten Bedingungen Genüge leistet. Bekanntlich hat schon Brunner über diesen Gegenstand interessante Mittheilungen gegeben und
einen Apparat dazu in Vorschlag gebracht. Ich habe die Einrichtung desselben
wesentlich abgeändert und sicherlich nicht auf Kosten der Genauigkeit noch der
Einfachheit, wie erhaltene Resultate mich bis jetzt belehrten, indem ich von der
Ansicht geleitet wurde den Apparat bei völligem Verschlüsse in Thätigkeit zu
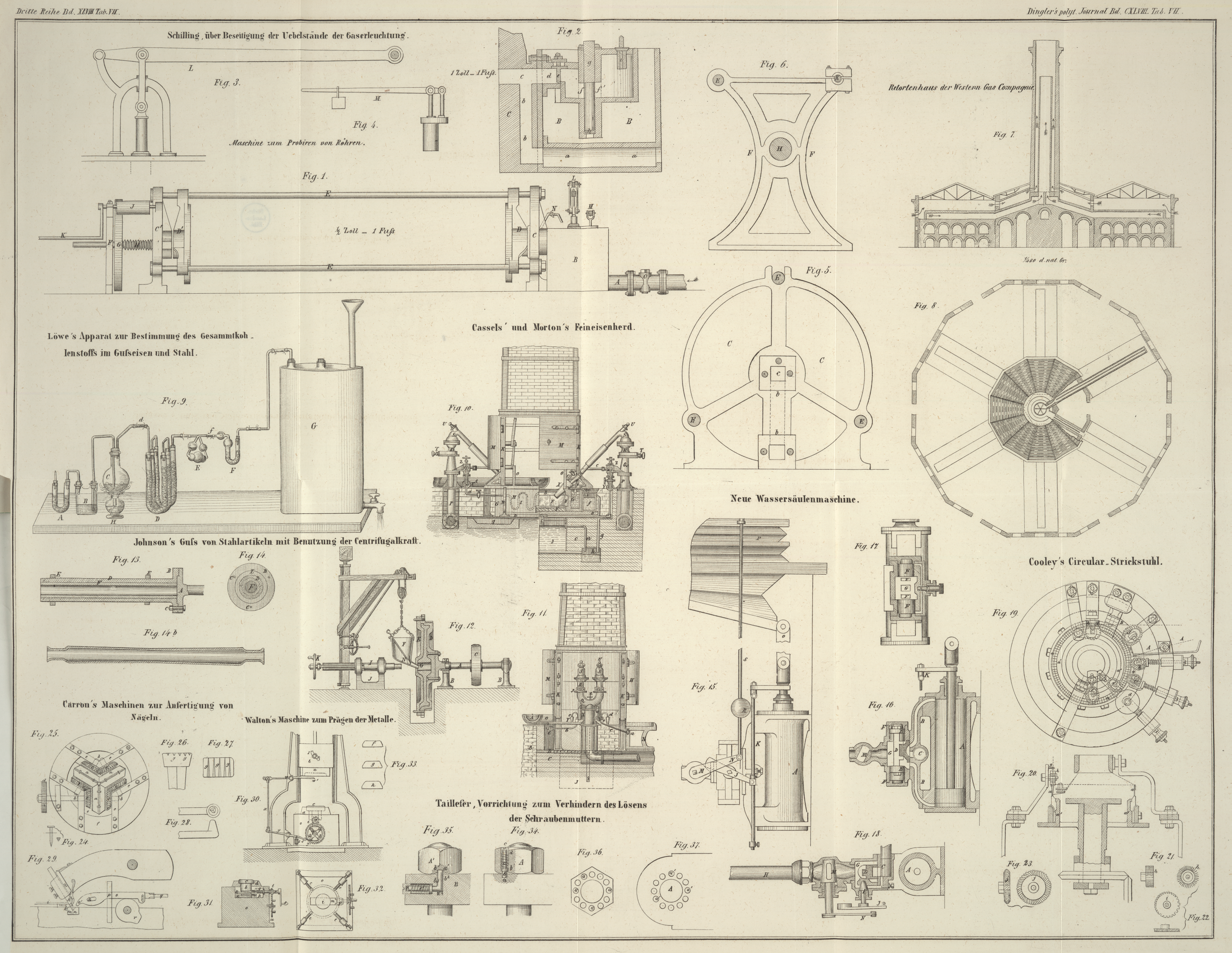
versetzen und die Oxydirung zu bewerkstelligen. Derselbe hat nach Fig. 9 auf Tab. VII
folgende Einrichtung: B ist eine kleine zweihalsige Woulf'sche Flasche, welche mit einer kalten Mischung von
2 Theilen reiner englischer Schwefelsäure und 1 Theil Wasser bis zu einem Drittheil
gefüllt ist. Ihr einer Tubulus ist mit einem durchbohrten Korke geschlossen, durch
welchen eine knieförmig gebogene mit der Uförmigen Röhre
A mittelst Korken verbundene Glasröhre geht. A ist mit festen Stückchen Aetzkali angefüllt, welch
letztere von den Korken mittelst lockeren Pfröpfchen von Asbest getrennt sind, und
erfüllt den Zweck die Kohlensäure der später durch den Apparat circulirenden Luft zu
binden. Der zweite Tubulus von B ist mit dem
durchbohrten Stopfen und der aus einem Stücke bestehenden
nicht zu engen und dünnen an beiden Enden offenen Glasröhre (so daß ein Abbrechen an
den Knieen dieser Röhre bei Bewegung des Apparates nicht zu befürchten steht), mit
dem Kölbchen C verbunden und endigt etwa 1 Linie über
dem Boden des letzteren. In diesem Kolben wird die Oxydation ausgeführt und aus
diesem Grunde wird in diesen vor der Operation der abgeschiedene Kohlenstoff nebst
der dazu nöthigen Menge fein zerriebenem reinem zweifach-chromsauren Kali
eingetragen. Das fest und luftdicht schließende Kölbchen hat eine zweite
Durchbohrung, durch welche die knieförmige etwas aufwärts gebogene unterhalb des
Stopfens endende offene Glasröhre d geht; sie ist
nämlich zu dem Zwecke aufwärts gebogen, damit die aus dem Kölbchen C beim Kochen verdampfende und sich abkühlende
Feuchtigkeit in dieses wieder zurückfließt. Letztere Röhre steht durch eine
Kautschukverbindung mit dem Röhrensystem D von Glas in
Vereinigung. Diese gebogenen Röhren sind mit erbsengroßen Stückchen porösen
Bimssteins gefüllt, deren Poren man vor dem Einfüllen sich mit reiner englischer
Schwefelsäure hat vollsaugen lassen. Ihre beiden Enden sind durch lockere vorher mit
Schwefelsäure und Wasser gewaschene und darauf geglühte Pfropfe von Asbest von den
mit der Säure getränkten Bimssteinstückchen getrennt und die ihre Mündungen
schließenden fehlerfreien Stopfen an ihrer inneren Seite noch mit Stanniol
überzogen, so daß eine
Verkohlung derselben nicht so leicht zu befürchten steht, im Falle etwas
Schwefelsäure sollte capillarisch von dem Asbeste
aufgesogen werden, oder an den inneren Wandungen der Glasröhren sich erheben. Ferner
werden die, die Röhren gut verschließenden Korkstopfen nach der Füllung der ersteren
noch mit einer Auflösung von Schellack in Weingeist überzogen; die diese letztere
durchdringenden und die drei gleichgroßen gefüllten Röhren verbindenden gebogenen
Glasröhren sind von mäßigem Durchmesser, jedoch stark im Glase und immer aus einem
Stücke, damit das ganze System Standfähigkeit besitzt. Die dritte der gebogenen
Röhren steht mittelst einer Kautschukverbindung mit dem Liebig'schen Kugelapparate E in Verbindung,
welch letzterer mit Kalilauge von der Stärke und Menge angefüllt ist, wie derselbe,
auf gleiche Art gespeist, Anwendung in der organischen Analyse findet. Sein zweites
Ende steht mit der gefüllten Chlorcalciumröhre F durch
Kautschuk in Verbindung, und diese letztere endlich mit dem geräumigen mit Wasser
völlig angefüllten Aspirator G. Diese letzte Röhre F hat den Zweck die Luft zu trocknen, welche durch den
Kugelapparat gegangen ist; bei der bekannten Stärke der Kalilauge des Kugelapparates
ist die Gewichtszunahme dieser nach Beendigung des Versuches höchst gering, sie ist
jedoch immer der größeren Genauigkeit halber dem Apparate einzuschalten. Es ist gut,
das mit der Chlorcalciumröhre in Berührung stehende Ende des Liebig'schen Kugelapparates vor der Lampe anfangs auszuziehen, damit es
sich in die Röhre F bei f
einschiebt und durch eine Kautschukverbindung gehalten und luftdicht verschlossen
wird. Geht dann vielleicht bei Beginn der Operation der Apparat etwas zu heftig und
sollte Kalilauge übergerissen werden, so fließt diese in die Kugel der Röhre F, ohne die Kautschukverbindung zu benetzen und ohne daß
somit ein Zweifel an der Genauigkeit des Versuches sich einschleichen kann oder gar
derselbe zu erneuern ist, was natürlich einen großen Zeitverlust im Gefolge hätte.
Bei einiger Vorsicht und Uebung steht dieser Zwischenfall jedoch nie zu befürchten. Denken wir uns den ganzen Apparat bis
zum Beginne der Operation fertig, so hat man nur nöthig den Hahn des Aspirators zu
öffnen und Wasser ausfließen zu lassen. Die Schwefelsäure der Flasche B wird auf diese Art in den Kolben C hinübergezogen, kommt hier mit der Kohle und dem
doppelt-chromsauren Kali in Berührung, macht aus letzterem die Chromsäure
frei, welche nun durch Sauerstoffabgabe oxydirend auf die Kohle einwirkt. Ist die
erste Einwirkung und Entbindung der Kohlensäure vorüber, so erwärmt man den Inhalt
des Kolbens C durch eine untergestellte kleine
Spiritus- oder Gasflamme H etwa 1 Stunde und läßt
immer durch Wasser-Ausfluß kohlensäurefreie Luft durch den Apparat circuliren. Die
gebildete Kohlensäure wird beim Durchgange durch das Röhrensystem D getrocknet und mechanisch durch den steten
Wasserausfluß in den Kalilauge haltenden Kugelapparat zur Bindung übergezogen. Den
Ausfluß des Wassers aus dem Hahne des Aspirators regulirt man so, daß in jeder
Secunde demselben ein Tröpfchen entfällt. Die Luft läßt man so mindestens 2 1/2
Stunden durch den ganzen Apparat strömen, damit man Sicherheit erhält, daß alle
Kohlensäure in E gebunden und ferner alles gleichzeitig
mit auftretende freie Sauerstoffgas durch die eingedrungene Luft aus den Theilen E und F verdrängt ist. Nach
Ablauf dieser Zeit werden die Enden der Röhren E und F mittelst gebundenen Kautschukstückchen verschlossen
und gewogen, der resultirende Gewichtsüberschuß nach Abzug der ersten von der
zweiten Wägung ist die Menge der Kohlensäure, aus welcher der Kohlenstoff mit großer
Genauigkeit zu berechnen ist. Es ist nothwendig zur Ausführung des Versuches, daß
der Aspirator sehr geräumig ist, indem sonst der Druck beim Ausflusse des Wassers zu
schwach wirkt, um das specifisch schwere Gemisch von Schwefelsäure und Wasser aus
der Flasche B durch die Röhre nach C überzuführen. Kennt man die Menge
zweifach-chromsauren Kalis, welche zur völligen Oxydation des Kohlenstoffs in
Wirkung kommen muß, so läßt sich annähernd auch die Menge der Schwefelsäure dem
Volumen nach angeben, welche zur Zersetzung hier nöthig, ohne daß man zu befürchten
hätte, ein kleiner Ueberschuß derselben in C wirke
nachtheilig. Man gibt daher der Röhre b vor Ausführung
der Operation eine solche Stellung, daß sie nicht tiefer in die Mischung der Flasche
B eintaucht, als dieses Volumen etwa beträgt, daß
sie also nach dessen Uebergang aus der hier überschüssig vorhandenen Flüssigkeit
tritt und das Kölbchen C sich durch das Uebergegangene
nicht weiter als bis höchstens zur Hälfte anfüllt. Geht man auf Grund der
vorliegenden Analysen von der Voraussetzung aus, daß die Menge des Kohlenstoffes im
Gußeisen im Mittel = 5 Proc., im Stahl dagegen 2 Proc. betrage, so würde man für je
1 Grm. Gußeisen und Stahl bei einem Kohlengehalte = 0,05 und = 0,02 sehr nahe 0,83
und 0,37 Grm. reines doppelt-chromsaures Kali nöthig haben, wenn 3 Aeq. Kohle
nach der Formel den Sauerstoff aus 2 Aeq. doppeltchromsauren Kali zur Oxydation und
Umbildung in 3 Aeq. Kohlensäure bedürfen. Jedenfalls ist es rathsam und für die
Genauigkeit der Analyse ohne Eintrag, wenn man die annähernd berechnete Menge des
rothen chromsauren Salzes verdoppelt, indem hier die Betrachtung von einer reinen
Kohle ausging, welches in der That nicht der Fall ist, da Wasserstoff,
ausgeschiedener Schwefel etc., wie schon im Laufe unserer Mittheilung erwähnt, hier ebenfalls
gegenwärtig sind und einen Theil des Sauerstoffs der freigewordenen Chromsäure für
sich in Anspruch nehmen.
Im Eingange dieser Mittheilung wurde vorgeschrieben, den mittelst Jod ausgeschiedenen
und gewaschenen Gesammtkohlenstoff genannter Eisensorten auf einem Papierfiltrum zu
sammeln. Diese Art der Aufsammlung ist natürlich hier unstatthaft, wo einen Theils
derselbe nach dem Trocknen und selbst feucht sich schwierig vollständig von dem
Papier wieder ablösen, und anderen Theiles selbst in diesem Falle nicht ohne die
Möglichkeit der Beimischung von Fasern des Papiers gewinnen ließe, wodurch im
Hinblick auf diese genannten Uebelstände der Kohlenstoffgehalt unbedingt zu niedrig
oder zu hoch ausfallen müßte. Am zweckmäßigsten erkannte ich nach mancherlei
Prüfungen ein Filtrum von Asbest, welch letzteren man durch Schwefelsäure und Wasser
gewaschen und darauf geglüht hierzu verwendet.
Textabbildung Bd. 148, S. 441
Derselbe wird in dem kegelförmigen Ausschnitt einer
Messing scheibe A locker ausgebreitet
und mittelst des aufgesetzten Metallconus B durch
gelindes Drücken und Drehen vereinigt. Auf diese Weise gelingt es leicht durch
einige Geschicklichkeit ein Filtrum zu formen, dessen offene Stellen und dessen
Spitze man leicht durch Anlegen von kleinen Mengen von Asbest verdichten kann.
Dieses so geschaffene Filtrum legt man fest in einen kleinen Glastrichter,
dessen Raum es nicht über die Hälfte anfüllt, und filtrirt erst die eisenhaltige
Lösung durch und sammelt zuletzt, was ohne den geringsten Verlust geschehen
kann, die ausgeschiedene Kohle auf demselben, die dann durch Waschen zu reinigen
ist. Nach Beendigung dieser Operationen wird der Glastrichter mit Papier zum
Schütze vor Staub überbunden und durch Einsehen in ein Becherglas, dessen Boden
abgesprengt, auf dem Sandbade getrocknet. Nach völligem Austrocknen läßt sich
dann dieses Asbestfiltrum leicht von dem Glastrichter abheben und in den Kolben
C ohne Verlust einführen. Sind etwa kleine
Mengen der bräunlichen Kohle an den Wänden des Glastrichters haften geblieben,
so wischt man diese mit gleichfalls gereinigten Asbestbäuschchen, welche man um
einen Glasstab legt, vorsichtig ab und trägt diese ebenfalls in den Kolben ein.
Dieses Aufsammeln der Kohle auf den Asbestfasern gewährt den Vortheil einer
größeren Vertheilung und schnelleren Oxydirung der organischen Substanz.
– Auch ist die Ausführung des Versuches nicht unterblieben, das fein
gefeilte, gebeutelte etc. kohlehaltige Eisen direct im Kolben C in gleicher Art zu behandeln, um ohne vorherige
Abscheidung des Kohlenstoffs gleich so dessen Procentgehalt zu ermitteln. Allein
es haben sich dadurch keinerlei Vortheile ergeben, im Gegentheil war hier ein
weit
größerer Aufwand an Zeit bis zur völligen Oxydation
erforderlich und die gefundenen Resultate meist zu niedrig – ein Uebelstanb
den schon Brunner in seiner Mittheilung hervorgehoben. Da
außerdem für diesen Fall der Zusatz des zweifach-chromsauren Kalis zu erhöhen
ist, so wird die Lösung einmal durch die Concentration der angewandten Säure, so wie
durch das vorhandene Eisensalz zu consistent und läßt sich ohne Mißstände nicht gut
erhitzen.
Bekanntlich hat auch Berzelius schon zur Abscheidung der
Kohle aus kohlehaltigen Eisensorten eine Lösung von Kupferchlorid vorgeschlagen.
Auch diese Methode hatte ich in Prüfung, als mir die obenerwähnte Arbeit von Max Büchner zukam, welcher sie ebenfalls empfiehlt und in
Anwendung brachte. Da es nothwendig, daß die für die Lösung des Eisens und
Bestimmung des Gesammtkohlenstoffs zum Angriff kommende Kupferlösung möglichst
neutral sey, und dieselbe sich ohne vorausgegangenes Abdampfen wegen des
Ueberschusses der Säure u.s.w., überhaupt ohne Zeitverlust für diesen speciellen
Fall nicht schnell in größerer Menge bereiten läßt, so nahm ich Rücksicht auf diesen
Fall und brachte eine Kupferchloridlösung in Anwendung, welche in kürzester Frist
und in großen Quantitäten neutral für unseren Zweck darzustellen ist. Ich stützte
mich dabei auf die Erfahrung von Rieckher, welcher
gefunden, daß eine wässerige Mischung von Kochsalz und Kupfervitriol beim Abdampfen
Glaubersalz, überschüssiges Kochsalz und zuletzt Krystalle von wasserhaltigem
Kupferchlorid gibt. Mit dieser Lösung ist der gewünschte Zweck unter demselben
Erfolge zu erzielen, wie mit der reinsten neutralen Kupferchloridlösung. Die
Bereitung derselben geschieht durch Verflüssigen eines Aequivalentes reinen
wasserhaltigen Kupfervitriols und eines Aequivalentes Kochsalz in möglichst wenig
Wasser, um die Mischung bei einem gewissen Volumen in etwas concentrirtem Zustande
zu besitzen, oder durch Auflösung von 2 Theilen krystallisirtem Kupfervitriol und 1
Thl. Kochsalz in Wasser. Man übergießt mit diesem wässerigen Gemische beider
genannten Salze, in welchem der Gehalt an schwefelsaurem Natron ohne Einfluß ist,
die in einem Becherglase befindliche abgewogene Eisenprobe, welche hierzu in dem
Grade der Vertheilung und Zerkleinerung ist, wie dieses bereits in dem Eingange
unserer Mittheilung für die Zersetzung mittelst Jods als nothwendig erkannt wurde.
In demselben Momente, wo die Kupferchloridlösung die seine Eisenfeile überlagert,
scheidet sich an letzterer rothes fein zertheiltes metallisches Kupfer aus, welches
erforderlich macht, daß man mittelst des Glasstabes die Probe umrührt, um das noch
ungelöst gebliebene metallische Eisen mit der Lösung wieder in Contact zu setzen,
weil sonst leicht der Rest der Eisenfeile durch eine Kupferhülle dem Angriffe der Lösung entzogen und
hierdurch die Zersetzung gehemmt oder wenigstens sehr verzögert wird. Sobald sich
beim Umrühren mit dem Glasstabe keine festen Körnchen am Boden des Glases mehr
fühlbar machen, ist die Operation beendet. Fast immer ist der rothe Schlamm von
metallischem Kupfer noch überlagert von einer weißen krystallinischen Rinde von
Kupferchlorür.
Man läßt die Flüssigkeit durch Absetzen sich klären, decantirt oder filtrirt sie von
dem ungelöst gebliebenen Rückstande ab und übergießt darauf letzteren mit einer
neuen Menge der genannten Kupferchloridlösung, welcher man nun einen größeren Zusatz
von etwas starker reiner Salzsäure gibt. Durch diese letztere Säure wird das
ausgeschiedene weiße Kupferchlorür sogleich aufgenommen und zum Verschwinden
gebracht. Man setzt das Becherglas nun in das heiße Wasserbad ein oder stellt es
einige Zeit auf das erwärmte Sandbad, wodurch das noch vorhandene rothe metallische
Kupfer ebenfalls unter Bildung von Kupferchlorür in die Lösung übergeht und nur die
Kohle als schwarzer Schlamm zurückbleibt. Diese ist auf dem Asbestfiltrum von
angegebener Art zu sammeln, zu waschen, zu trocknen und in dem verzeichneten
Apparate, wie bereits erwähnt, zu oxydiren. Die Aufnahme des beim
Zersetzungsprocesse ausgeschiedenen metallischen Kupfers durch das Kupferchlorid und
die Salzsäure erfolgt etwas träge, wenn man nicht die Operation durch anhaltendes
Erwärmen auf mindestens 70° C. unterstützt und öfters den Satz mit dem
Glasstabe aufrührt.
Der in unserer Mittheilung angegebene Apparat zur Bestimmung des Gesammtkohlenstoffs
im Gußeisen und Stahl ist jedoch noch anderer als nur dieser speciellen Anwendung
fähig, und läßt sich in der Praxis, wo es sich um Erreichung nur annähernder
Resultate handeln sollte, mannichfach benützen, wie z.B. bei Ermittelung des
theoretischen Heizvermögens verschiedener fossilen Kohlen und anderer ähnlichen
Heizmaterialien, sobald natürlich der Gehalt derselben an Wasserstoff und Sauerstoff
das Resultat in besonderen Fällen als nicht alterirend angesehen werden darf; ferner
zur Werthbestimmung des Graphites u. dgl. Er gewährt den Vortheil einer großen
Zeitersparnis indem mehrere solcher Analysen sich nacheinander ausführen lassen,
ohne daß besondere Vorbereitungen dafür nothwendig. Ist die Kalilauge des
Kugelapparates von der richtigen Concentration, so läßt sie sich ohne Erneuerung zu
einer größeren Anzahl von Versuchen anwenden, und bei einem guten sorgfältigen
Verschlüsse ihrer Röhren kann selbst nach 24 Stunden die letzte Wägung einer neuen
Bestimmung zu Grunde gelegt werden, was natürlich die auf die Analysen zu
verwendende Zeit sehr abkürzt.
Zur Prüfung des Apparates wurde bis jetzt öfters unkrystallisirte und durch scharfes
Pressen zwischen Fließpapier getrocknete Oxalsäure von der Formel C² O³ 3HO
angewandt und deren Kohlenstoff in Procenten ermittelt.
Es wurde hiernach
Textabbildung Bd. 148, S. 444
Diese Versuche werden noch für andere organische Substanzen, wie Zucker, Stärkmehl u.
dgl. fortgesetzt, um zu bestimmen, ob die Ermittelung des Kohlenstoffgehalts dieser
mit derselben Genauigkeit sich hierdurch erzielen läßt.
Tafeln