| Titel: | Chemische Mittheilungen; von Prof. C. Brunner. |
| Fundstelle: | Band 150, Jahrgang 1858, Nr. XCII., S. 369 |
| Download: | XML |
XCII.
Chemische Mittheilungen; von Prof. C. Brunner.
Vorgetragen in der schweizerischen
naturforschenden Gesellschaft am 23. Oct. 1858. – Aus den Berner Mittheilungen,
Nr. 417 und 418.
Mit Abbildungen auf Tab.
VI.
Brunner's chemische Mittheilungen.
1. Trennung von Zink und
Nickel.
Zur Trennung und quantitativen Bestimmung von Zink und Nickel sind in neuerer Zeit
mehrere Methoden empfohlen worden. Eine der einfachsten scheint die von Smith angegebene zu seyn. Dieselbe gründet sich auf den
Umstand, daß aus einer essigsauren Lösung beider Oxyde durch Schwefelwasserstoffgas
nur das Zink gefällt wird.
Bei diesem Verfahren macht RoseHandbuch der analytischen Chemie, Bd. II S. 65. die Bemerkung, daß nur dann eine genaue Trennung erfolge, wenn in der
Flüssigkeit keine starke Säure, nur Essigsäure, vorhanden sey.
RammelsbergAnfangsgründe der quantitativen Analyse, S. 78. erklärt die Methode für ungenau und sagt ausdrücklich, daß mit dem Zink
immer Nickel niedergeschlagen werde.
Eine Reihe von Versuchen, welche die einzelnen bei diesem Verfahren vorkommenden
Umstände zum Gegenstand hatten, führte zu einer Operationsmethode, die ein
zuverlässiges Resultat zu geben scheint.
Man stellt zuerst die beiden Metalle als salzsaure oder salpetersaure Auflösung dar,
die man so weit verdünnt, daß auf 1 Gramm beider Oxyde wenigstens 500 Gram.
Flüssigkeit kommen, sättigt nun diese annähernd mit kohlensaurem Natron, so daß nur
eine sehr geringe Menge von freier Säure zugegen bleibt. Um diesen Punkt genau zu
treffen, fügt man so lange eine verdünnte Lösung des Natronsalzes hinzu, bis nach
einigem Umschütteln und Stehenlassen der Niederschlag nicht völlig verschwindet,
worauf man ihn durch einige Tropfen Säure fortnimmt. Man leitet nun
Schwefelwasserstoffgas durch die Flüssigkeit, wodurch nach einiger Zeit ein
vollkommen weißer Niederschlag (Schwefelzink) entsteht. Nachdem ein guter Antheil
Zink auf diese Weise gefällt worden, setzt man der Flüssigkeit einige Tropfen einer
sehr verdünnten Lösung von essigsaurem Natron zu, und fährt fort Schwefelwasserstoff
durchzuleiten, so lange als sich der Niederschlag zu vermehren scheint, und läßt
hierauf die Flasche 10–12 Stunden bei gewöhnlicher Temperatur stehen. Der
Niederschlag senkt sich vollkommen, und kann sehr gut auf dem Filter gewaschen
werden.
Um sich zu versichern, daß alles Zink gefällt sey, wird eine Probe der filtrirten
Flüssigkeit mit 1 Tropfen verdünnter Lösung von essigsaurem Natron versetzt und mit
Schwefelwasserstoff behandelt. Sollte noch eine weißliche Trübung entstehen, so
müßte die ganze Flüssigkeit ebenso behandelt werden.
Aus der nunmehr von Zink befreiten Flüssigkeit kann nun das Nickel nach Austreiben
des Schwefelwasserstoffes durch Erwärmung, mittelst Kalihydrat gefällt werden. Der
Niederschlag von Schwefelzink wird, nach gehörigem Auswaschen, mit dem Filter in ein
Glas gegeben, mit Salzsäure digerirt, bis aller Geruch von Schwefelwasserstoff
verschwunden ist, die mit Wasser verdünnte Lösung filtrirt und das Zink nach den
bekannten Methoden bestimmt.
Bei dieser Scheidung spielt das essigsaure Natron offenbar eine vermittelnde Rolle.
Es entsteht nämlich durch Umsetzen eine kleine Menge essigsaures Zinkoxyd, welches
durch den Schwefelwasserstoff gefällt wird. Die freigewordene Essigsäure bildet von
neuem essigsaures Zinkoxyd, welches sofort wieder gefällt wird. Es dürfte die Wirkung mit der
Bildung von kohlensaurem Bleioxyd durch Einwirkung von kohlensaurem Gase auf ein mit
Wasser angerührtes Gemenge von Bleiglätte und Bleizucker zu vergleichen seyn. Es ist
daher begreiflich, warum eine nur so höchst geringe Menge von essigsaurem Natron
erforderlich ist.
Damit die Scheidung genau sey und kein Nickel mit dem Zink gefällt werde, sind
folgende Cautelen zu beobachten:
1) Die Lösung muß anfänglich ein wenig, doch nur sehr schwach, sauer seyn; ich möchte
sagen 1–2 Tropfen freie Säure enthalten. Ist sie vollkommen neutral, so
erscheint der Niederschlag durch Schwefelwasserstoff schmutzig gefärbt,
nickelhaltig. Ist das Verhältniß richtig getroffen, so ist er rein weiß. Nach dem
Auswaschen kann dann weder durch das Löthrohr noch auf andere Art Nickel darin
gefunden werden.
2) Eine zu große Menge essigsaures Natron, sowie auch jede Erwärmung muß vermieden
werden. Setzt man nämlich eine etwas bedeutende Menge essigsaures Natron hinzu, so
fällt etwas Nickel nieder, ja man kann hiedurch, besonders wenn zugleich erwärmt
wird, alles Nickel vollständig niederschlagen.
Bei Versuchen mit genau abgewogenen Mengen von Oxyden (0,2 bis 0,3 Gramm eines jeden)
wurden dieselben bis auf 1–2 Milligramme wieder erhalten.
Auf die nämliche Art kann Zink von Kobalt getrennt werden.
Das aus einer kobalthaltigen Lösung abgetrennte Schwefelzink gab stets ein Oxyd,
welches vor dem Löthrohr mit Borax keine Färbung hervorbrachte.
Endlich ist noch zu bemerken, daß wenn Eisen zugegen ist,
dieses vorher abgeschieden werden muß, indem es sonst theils in den Zink-,
theils in den Nickelniederschlag eingeht. Für diesen Fall paßt am besten die
bekannte Fuchs'sche Methode mit kohlensaurem Baryt und
nachheriges Entfernen des Baryts durch Schwefelsäure. Die Abscheidung mit Ammoniak
ist nicht anwendbar, da hiedurch die nachherige Trennung der beiden Metalle
unmöglich würde.
2. Einwirkung von Ammoniakflüssigkeit
auf Schwefel.
Es kommt nicht selten vor, daß man über die gewöhnlichsten Dinge in unsern
Handbüchern keinen Aufschluß findet. So z.B. wird man umsonst über das Verhalten der
Ammoniakflüssigkeit (Salmiakgeist) gegen Schwefel Belehrung suchen. Nur bei Rose
Handbuch der analytischen Chemie, Bd. I S. 433. finde ich die Angabe, daß Ammoniakflüssigkeit reinen Schwefel nicht auflöse, wohl
aber arsenikhaltigen. Ein specieller Fall veranlaßte mich, diesen Gegenstand näher
zu untersuchen. Das Ergebniß war folgendes:
Digerirt man reinenEs wurde sicilianischer Schwefel durch Destillation gereinigt und nach
Zerreiben mit destillirtem Wasser so lange ausgekocht, bis das Wasser nicht
mehr mit Chlorbaryum reagirte. Schwefel mit Ammoniakflüssigkeit, so wird, wenn die Temperatur nicht
60° R. übersteigt, selbst nach längerer Zeit keine Einwirkung wahrgenommen.
Wird jedoch die Flüssigkeit stärker erwärmt, etwa auf 70°, so nimmt sie eine
schwach gelbliche Färbung an, welche beim Kochen noch deutlicher hervortritt. Es hat
sich nun eine sehr kleine Menge von Schwefel aufgelöst, denn die Flüssigkeit gibt
mit essigsaurem Bleioxyd einen bräunlichrothen Niederschlag. Schwefelsäure enthält
sie nicht. Sättigt man eine Probe mit Salzsäure und filtrirt den niedergeschlagenen
geringen Schwefelniederschlag ab, so gibt Chlorbaryum selbst nach längerer Zeit
nicht die geringste Trübung.
In einer gut verschlossenen Flasche läßt sich die Lösung von Schwefel in Ammoniak
unverändert aufbewahren. Selbst nach einigen Wochen ist dieselbe noch gelblich
gefärbt und vollkommen klar, gibt auch mit Bleisolution den röthlichen Niederschlag.
Bei Zutritt von atmosphärischer Luft trübt sie sich bald. Nach 24 Stunden hat sich
ein geringer Schwefelniederschlag gebildet. Die von demselben abfiltrirte Lösung
gibt nun mit Bleisolution einen weißen Niederschlag, mit Chlorbaryum eine sehr
geringe Reaction auf Schwefelsäure.
Kocht man den nämlichen Schwefel wiederholt mit Ammoniakflüssigkeit, so nimmt er eine
blasse, etwas ins Grauliche spielende Färbung an. Wird dieses so oft wiederholt, bis
der meiste Schwefel aufgelöst ist, so bleibt ein flockiger grauschwarzer Rückstand,
der beim Erhitzen mit doppeltchromsaurem Kali und Schwefelsäure vollkommen
verschwindet. Es ist dieses offenbar ein wenig Kohle, die in allem, selbst durch
zwei- bis dreimalige Destillation gereinigtem Schwefel, enthalten zu seyn
scheint.
3. Bereitung des molybdänsauren
Ammoniaks.
Seitdem die Anwendung dieses Salzes zur Entdeckung der Phosphorsäure für die
chemische Analyse unentbehrlich geworden ist, wurden mehrere Methoden zu seiner
Darstellung angegeben. Die meisten gehen darauf hinaus, den natürlichen
Molybdänglanz bei Luftzutritt so lange zu rösten, bis aller Schwefel verbrannt und
das Molybdän in Molybdänsäure verwandelt ist, die nachher in Ammoniakflüssigkeit
gelöst wird. Diese Operation wird gewöhnlich in einem schief liegenden Platintiegel
unter öfterem Umrühren der Masse vorgenommen. Man wird wohl allgemein hierbei die
Erfahrung gemacht haben, wie langwierig es ist, sie zu Ende zu führen. Die kürzlich
von Wöhler
Annalen der Chemie und Pharmacie, Bd. C S. 376. angegebene Verbesserung dieses Verfahrens durch Anwendung eines mittelst des
Aspirators hervorgebrachten Luftzuges führt ebenfalls nur langsam zum Ziel. Der
Grund hievon liegt theils in dem Umstande, daß es schwer hält das Material
hinlänglich zu zertheilen, da es durch Anwendung der Wärme immer wieder
zusammenbackt; theils darin, daß die entstehende Molybdänsäure das noch übrige
Mineral bedeckt und dadurch seine Verbrennung erschwert.
Auf folgende Art gelingt die Operation sehr leicht: Man reibt den Molybdänglanz mit
ungefähr seinem gleichen Volumen groben mit Salzsäure gewaschenen Quarzsandes in
einer Achatschale zu mäßig feinem Pulver, gibt dieses auf eine flache Platinschale
oder Platinblech, und erhitzt es über einer guten Weingeistlampe unter öfterem
Umrühren zum anfangenden Glühen, so lange bis das Gemenge eine citrongelbe (nach dem
Erkalten weißliche) Farbe angenommen hat. Eine Viertelstunde ist hiezu für eine
Menge von einigen Grammen vollkommen ausreichend. Nach dem Erkalten wird die Masse
mit Ammoniakflüssigkeit ausgezogen und auf die bekannte Art weiter behandelt.
4. Bestimmung der Niederschläge bei
Analysen.
Wir verdanken bekanntlich Berzelius die jetzt allgemein
übliche Methode die Niederschläge bei chemischen Analysen mit dem Filter zu glühen,
und ihre Menge durch directe Wägung mit Abzug der Asche des Filters zu bestimmen. So
einfach dieses Verfahren ist, so kommen doch zuweilen zwei Unbequemlichkeiten dabei
vor. Die eine ist die oft etwas langwierige gänzliche Verbrennung selbst bei
Anwendung der bekannten Handgriffe; die andere betrifft die bei einigen
Niederschlägen durch die Kohle des Filters anfänglich eintretende Reduction, wobei
sich das reducirte Metall stellenweise mit dem Platin des Tiegels legirt. Glüht man
z.B. einen Niederschlag von Zinkoxyd mit dem Filter, so wird man am Tiegel deutliche
Flecken dieser entstandenen Legirung wahrnehmen. Sind diese zwar von keinem
quantitativen Belang, und können sie mit Salzsäure leicht entfernt werden, so ist es
doch wünschenswerth diesen Umstand, der sich noch auf andere Niederschläge
erstrecken mag, zu vermeiden.
Folgende Methode hat sich seit längerer Zeit bestens bewährt:
Als Gefäß worin die Niederschläge geglüht werden, dient eine ungefähr 15 Centimeter
lange und 12 Millimeter weite Röhre von böhmischem Glase (von der Art, wie sie zu
Elementaranalysen benutzt werden). Dieselbe ist an dem einen Ende zu einer nicht
ganz feinen Spitze ausgezogen, in welche ein wenig Amianth leicht eingesteckt wird.
So vorgerichtet wird sie nebst einem Gewichtstück, welches das Gewicht des zu
bestimmenden Niederschlages um etwas übertrifft, auf der Waage aufs Genaueste
tarirt. Alsdann wird das mäßig getrocknete Filter mit dem Niederschlag
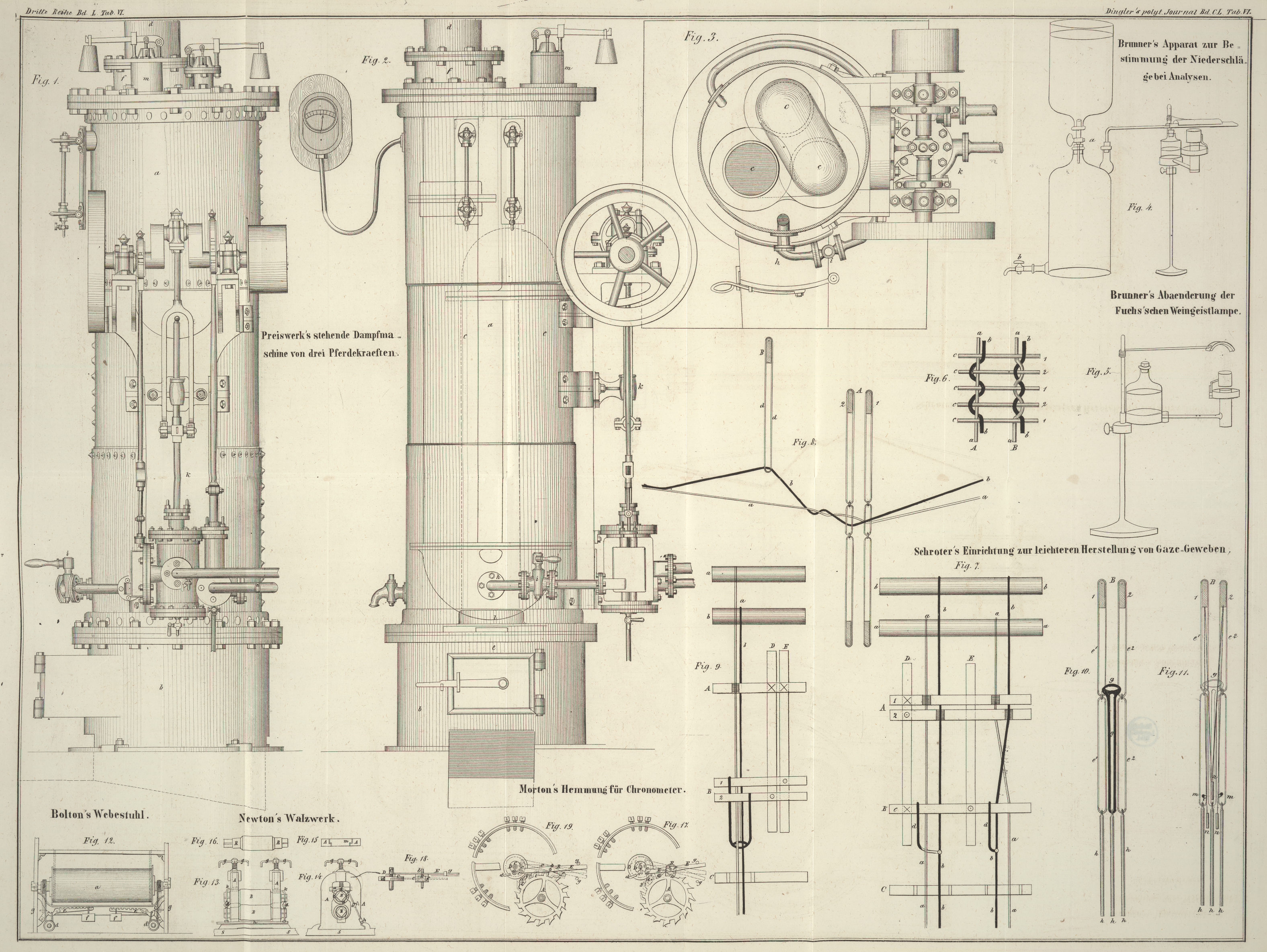
zusammengerollt in die Röhre hineingeschoben, und diese, wie Fig. 4 zeigt, mit einer
Flasche von etwa 3–4 Liter verbunden. Man läßt nun aus dem über der Flasche
angebrachten Gefäß durch Oeffnen des Hahnen a Wasser in
dieselbe fließen, so daß die atmosphärische Luft durch die Verbrennungsröhre
getrieben wird, während man zu gleicher Zeit diese letztere mittelst einer
Weingeistlampe mit doppeltem Luftzuge erhitzt. Die empyreumatischen Producte die das
Filter liefert, treten in Form eines Rauches aus der Spitze der Verbrennungsröhre
herausWill man die Unbequemlichkeit dieses Rauches vermeiden, so kann man ihn durch
eine vor die Oeffnung gestellte kleine Weingeistlampe verbrennen lassen., später verkohlt und verbrennt das Filter vollständig. Zuweilen ist es gut,
durch einige leichte Schläge an die Röhre den Inhalt derselben etwas zu zertheilen.
Man wird immer finden, daß die Verbrennung sehr leicht und vollständig erfolgt.
Nach Erkalten des Apparates wird die Röhre wieder auf die Waage gebracht, das
mittarirte Gewichtstück durch die erforderlichen Gewichte ersetzt, und so die Menge
des Niederschlages bestimmt, wobei das Gewicht der Filterasche in Abzug zu bringen
ist.
Es ist leicht einzusehen, daß die Operation nicht mehr Zeit erfordert, als die
gewöhnliche durch Glühen im Tiegel. Zwei Wägungen und eine Verbrennung sind bei
beiden erforderlich. Letztere geht in der Regel rascher als im Tiegel von statten.
Es könnten Manche sich durch den anzuwendenden Apparat abschrecken lassen. Hierauf
bemerke ich, daß ein solcher bleibend aufgestellt keine weitere Mühe veranlaßt. Man
kann seine Anwendung noch dadurch vereinfachen, daß, wenn die Flasche mit Wasser
gefüllt ist, man den Hahnen a schließt und durch Oeffnen
des untern b das Wasser in ein untergestelltes Gefäß
abfließen läßt. In diesem Falle entsteht ein Luftzug in entgegengesetzter Richtung,
der ebenso wie jener benutzt wird. Da bei diesen Versuchen stets Glühhitze angewandt
wird, so ist es überflüssig die Luft zu trocknen. Sollte man den Apparat zu anderen
Zwecken anwenden, z.B. zum Austrocknen, so müßte der Luftstrom durch eine mit
Bimsstein und Schwefelsäure versehene Röhre geleitet werden.
Noch muß ich einer kleinen Vorrichtung erwähnen, welche bei allen solchen Arbeiten
durch Erhitzung in Röhren von größtem Vortheil ist. Man bringt nämlich oberhalb der
zu erhitzenden Glasröhre einen gewölbten Reflector an, welcher den Zweck hat, die
Röhre von oben zu erwärmen. Da derselbe länger ist als die durch die Lampe erhitzte
Stelle, so erwärmt er zugleich die Röhre zum voraus, ehe die Lampe an die zu
glühende Stelle gelangt. Hiedurch wird alles Wasser weit vor der Lampe hergetrieben.
Da dieser Reflector, der aus einem Blatt von Schwarzblech gemacht ist und sich
mittelst eines spiralförmig gewundenen Drahtes an dem Ständer der Lampe verschieben
oder auch entfernen läßt, sich mitten über der Lampe befindet und immer mit
derselben weiter geschoben wird, so wird die Flamme, selbst bei Anwendung längerer
Röhren, nie an eine Stelle gelangen, wo sich Wasser befindet. Mit einiger Uebung
wird man bald dahin gelangen, daß niemals eine Röhre reißt. Nur ist zu empfehlen,
derselbe, eine ganz kleine, kaum merkliche Neigung nach vorn, d.h. nach dem noch zu
erhitzenden Theile, zu geben.
Bei dieser Gelegenheit erlaube ich mir eine Abänderung der Fuchs'schen Lampe zu beschreiben, deren Anwendung sich seit vielen Jahren
bewährt hat.
Fig. 5 ist
eine gläserne Flasche von beiläufig 180 Grammen Wassergehalt. Der Boden derselben
ist durch eine messingene mit der Flasche verkittete Kapsel ersetzt, aus welcher die
Ausflußröhre nach dem Argand'schen Brenner führt. Dieser ist nahe an seinem obern
Ende mit einer messingenen Kapsel umgeben, in welche Wasser gegossen wird. Hiedurch
wird verhindert, daß bei länger anhaltendem stärkeren Brennen der Weingeist ins
Kochen gelangt.
Die Flasche wird nicht, wie es gewöhnlich geschieht, durch eine Stellschraube
unmittelbar an dem Ständer befestigt, sondern ruht auf einem in einer Hülse am
Ständer leicht verschiebbaren, hölzernen durchbohrten Cylinder, welcher mit einer
Stellschraube versehen ist.
Hiedurch wird der Vortheil erlangt, daß die Lampe durch schnelles Drehen plötzlich
unter dem Apparate, auf den sie einwirkt, entfernt werden kann, welches in manchen
Fällen sehr erwünscht seyn kann.
5. Bereitung von kohlensaurem
Baryt.
Die gewöhnliche Bereitung dieses Salzes zu chemischem Gebrauche durch Niederschlagen
einer Auflösung von Chlorbaryum mit kohlensaurem Natron oder Ammoniak ist zwar
ganz rationell, und liefert ein vollkommen reines Präparat. Nur ist das vollständige
Auswaschen des Niederschlages etwas zeitraubend. Dieses wird auf folgende Art
abgekürzt:
Man macht ein Gemenge von 2 Th. krystallisirtem Chlorbaryum und 1 Th. wasserfreiem
kohlensaurem Natron,Die genaue Berechnung zu gleichen Aequivalenten würde auf 100 Chlorbaryum
43,3 kohlensaures Natron verlangen. Ein geringer Ueberschuß des letztern ist
jedoch von keinem Nachtheil. setzt noch 2 Th. Kochsalz hinzu und bringt das Gemenge in einem Thon-
oder bei kleinen Quantitäten in einem Platintiegel zu mäßigem Glühen. Nach dem
Erstarren wird die Masse in einer Schale mit Wasser übergossen. Nach 24 Stunden hat
sie sich vollkommen aufgeweicht. Der als feinkörniges Pulver ausgeschiedene
kohlensaure Baryt kann sehr leicht ausgewaschen werden.
Der Zusatz von Kochsalz gewährt den Vortheil, daß das nachherige Ausziehen mit Wasser
dadurch sehr erleichtert wird. Wird derselbe weggelassen, so bildet die Mischung
nach dem Glühen eine harte feste Masse, welche vom Wasser nur sehr schwer
angegriffen wird.
Auf eben dieselbe Art kann durch Glühen von 2 schwefelsaurem Zinkoxyd und 1
wasserfreiem kohlensaurem Natron reines Zinkoxyd bereitet werden. Hiebei ist ein
Zusatz von Kochsalz unnöthig.
6. Bereitung von
Platinschwarz.
Wir besitzen viele Methoden zur Darstellung des Platins in demjenigen Zustande, den
man seiner schwarzen Farbe wegen mit dem Namen Platinschwarz oder Platinmohr zu bezeichnen
pflegt. Bei den meisten neuern Bereitungsarten werden organische Substanzen,
Alkohol, Zucker u. dgl. als Reductionsmittel angewandt, wobei immer einiger Zweifel
übrig bleibt, ob nicht eine, vielleicht sehr geringe Menge organischer Substanz dem
Präparat anhänge.
Auf folgende Art erhält man ohne Anwendung organischer Substanzen sehr leicht einen
vollkommen reinen Platinmohr:
Man erhitzt in einer flachen Schale trockenes oxalsaures Eisenoxyd (durch
Niederschlagen von Eisenvitriol mit Oxalsäure bereitet und gehörig ausgewaschen) bis
zum anfangenden Verglimmen, setzt alsdann unter Umrühren die Erhitzung fort, bis
sich das Salz vollständig in Oxyd verwandelt hat. Das so dargestellte höchst feine
Pulver wird in einer Glasröhre bei einer kaum zum anfangenden Glühen gesteigerten
Temperatur durch einen
Strom trockenen Wasserstoffgas reducirt.Diese Reduction kann auf einer Weingeistlampe mit doppeltem Luftzuge, unter
Anwendung der oben (unter Nr. 4) beschriebenen Vorrichtung, geschehen. Nach gänzlichem Erkalten im Gasstrom schüttet man das zuweilen pyrophorische
Präparat in eine Schale mit Wasser und zerdrückt es darin mit einem Pistill durch
gelindes Reiben. Man trägt nun von diesem mit Wasser angerührten metallischen Eisen
so lange kleine Portionen in eine verdünnte, mit einem geringen Ueberschuß von
Salzsäure vermischte Lösung von Platinchlorid, bis diese nach kräftigem Schütteln
und einigem Hinstellen gänzlich entfärbt erscheint. Der erhaltene Niederschlag wird
nun nach Abgießen der Flüssigkeit zu wiederholten Malen mit concentrirter
Salpetersäure gekocht, bis der letzte Auszug keine bemerkenswerthe Menge Eisen
enthält, zuletzt die anhängende Salpetersäure durch eine schwache Kalilösung
entfernt.
Das so dargestellte Präparat erscheint als ein amorphes schwarzes Pulver; durch
Reiben in einer Achatschale nimmt es eisenartigen Glanz an. Beim Erhitzen in einem
Platinlöffel kommt es bei etwa 200° C. plötzlich ins Glühen und verwandelt
sich unter Verdoppelung seines Volumens in die gewöhnliche Form, dem Platinschwamm
ähnlich. Mit einem Tropfen Alkohol befeuchtet, geräth es ebenfalls nach 1–2
Secunden ins Glühen unter Verwandlung in die gewöhnliche Form.
Es leidet wohl keinen Zweifel, daß dem Präparate alle übrigen vom Platinschwarz
bekannten Eigenschaften zukommen werden. Sollte jemals von diesem Anwendung gemacht
werden, so dürfte sich obige Bereitung ihrer Einfachheit wegen empfehlen.
7. Bestimmung des Kohlengehaltes der
Kalksteine.
Es kann vielleicht bisweilen von geologischem Interesse seyn, den Kohlengehalt der
Kalksteine zu bestimmen. Die folgende Methode gründet sich auf den bekannten
Umstand, daß der Kohlenstoff durch die gleichzeitige Einwirkung von chromsaurem Kali
und Schwefelsäure in Kohlensäure verwandelt wird. Das Verfahren ist folgendes;
Eine gewogene Menge des zu untersuchenden Gesteins wird in erbsengroße Stücke
zerschlagen mit verdünnter Salzsäure behandelt, mit der Vorsicht, daß ein guter
Ueberschuß dieser letzteren angewendet und die Flüssigkeit zuletzt erhitzt wird. Die
Auflösung wird mit diesem Rückstande in ein Cylinderglas gegossen und nach Absetzen
des Ungelösten dieses durch mehrmaliges Decantiren ausgewaschen. Hierauf spült man
den Rückstand in ein Kochglas und setzt etwas Schwefelsäure hinzu. Man nimmt auf 100 Gramme des den
Rückstand bedeckenden Wassers ungefähr 15 Gramme Schwefelsäure. Die Flasche wird nun
mit einer Gasröhre versehen, deren zweiter absteigender Schenkel in eine kleine
Flasche taucht, welche eine klare Mischung von Chlorbaryumlösung und Ammoniak
enthält und zur Abkühlung in einem Gefäße mit Wasser steht. Man bringt nun zum
Kochen. Sollte sich in der vorgesetzten Flasche eine merkliche Trübung bilden,
welche auf einen Rückhalt von Kohlensäure schließen ließe, so wird das Kochen so
lange fortgesetzt, bis eine neue Probe der vorgeschlagenen Flüssigkeit nicht mehr
getrübt wird. Man bringt nun in die Kochflasche 2–3 Gramme
doppelt-chromsaures Kali in Krystallen, setzt von Neuem die Gasröhre ein und
läßt die Flüssigkeit wenigstens eine halbe Stunde lang anhaltend kochen. Die
entwickelte Kohlensäure wird nun als kohlensaurer Baryt in der vorgesetzten Flasche
erhalten.
Um die Menge des Niederschlages zu bestimmen, wird die Flasche nach Beendigung der
Operation sorgfältig verschlossen so lange hingestellt, bis sich derselbe vollkommen
zu Boden gesetzt hat, dann mehrmals durch Decantation, zuletzt auf dem Filter,
ausgewaschen, getrocknet und geglüht.
Wenn die Operation richtig ausgeführt wurde, so bleibt in der Kochflasche entweder
gar kein ungelöster Rückstand oder wenigstens, was der gewöhnliche Fall ist, ein
solcher, dessen Farbe und Ansehen keinen Kohlengehalt mehr annehmen läßt. Sollte man
hierüber in Zweifel seyn, so kann die Flüssigkeit noch einmal gekocht und das Gas in
eine neue Probe von Chlorbaryum und Ammoniakflüssigkeit geleitet werden.
Da bei diesen Untersuchungen gewöhnlich ein sehr geringer Gehalt von Kohle gefunden
wird, etwa 1/1000 und noch weniger, so ist anzurathen etwas größere Mengen des
Materials, etwa 100 Gramme, in Arbeit zu nehmen.
8. Reinigen von Gläsern und
Schalen.
Nicht selten kommt man in Verlegenheit, wenn Gläser oder Porzellanschalen, an denen
sich organische Stoffe festgesetzt hatten und durch die Länge der Zeit so
festgetrocknet sind, daß sie allen Auflösungsmitteln widerstehen, gereinigt werden
sollen. Folgendes Verfahren wird in beinahe allen Fällen ausreichen:
Man befeuchtet die zu reinigenden Stellen mit concentrirter Schwefelsäure, streut
hierauf zerriebenes doppelt-chromsaures Kali auf die Säure und läßt den
Gegenstand einige Stunden (etwa über Nacht) an einem mäßig warmen Orte stehen. Alle
organischen Stoffe werden hiedurch zerstört unter Bildung von schwefelsaurem
Chromoxyd, welches nebst der noch übrigen Säure durch Wasser entfernt wird.
9. Reinigen der Malerpinsel von
eingetrockneten Oelfarben.
Auf öftere Anfragen von Malern nach einem hiezu geeigneten Mittel, stellte ich eine
Reihe von Versuchen an, aus denen folgende Reinigungsmethode hervorging.
Man bereitet eine Lösung von 1 krystallisirtem kohlensauren Natron in 3 Wasser, hängt
die zu reinigenden Pinsel so in diese in einem Cylinderglase (Trinkglase) enthaltene
Lösung, daß sie etwa 2 Zoll von dem Boden des Glases entfernt bleiben, und läßt den
Apparat bei gelinder Wärme (60–70° C.) 12–24 Stunden stehen.
Selten wird eine längere Einwirkung erforderlich seyn. Die eingetrocknete Farbe ist
nun so weit aufgeweicht, daß sie mit Leichtigkeit auf die bekannte Art mit Seife
weggebracht werden kann. Steinhart vertrocknete Pinsel wurden durch dieses Verfahren
wieder brauchbar gemacht.
Wesentlich ist es, die angegebene Temperatur nicht zu überschreiten, da sonst die
Haare, besonders der Borstenpinsel, angegriffen und gänzlich verdorben werden.
Tafeln