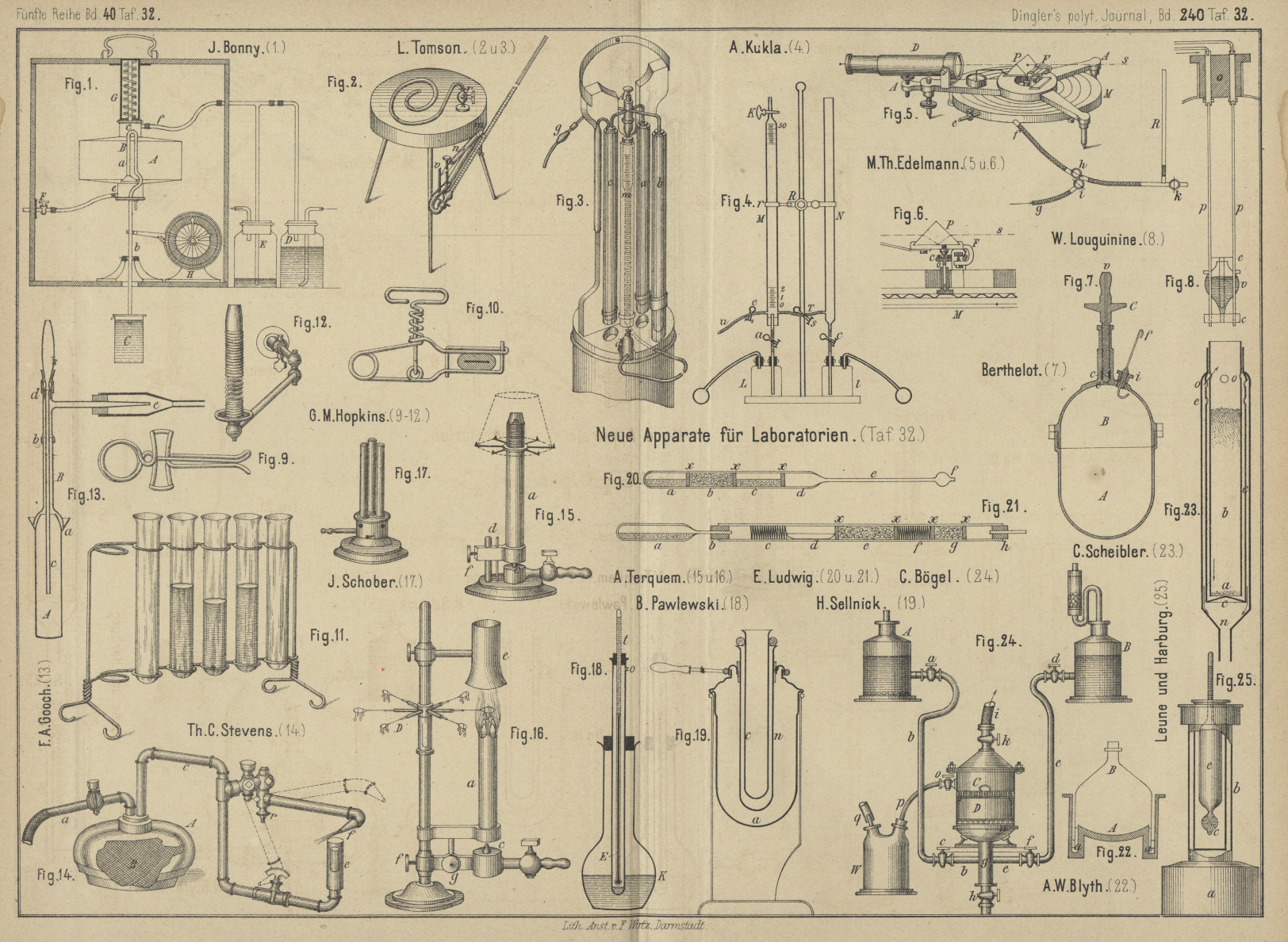
| Titel: | Neuere Apparate für Laboratorien. |
| Fundstelle: | Band 240, Jahrgang 1881, S. 373 |
| Download: | XML |
Neuere Apparate für Laboratorien.
Mit Abbildungen auf Tafel 32.
Neuere Apparate für Laboratorien.
Einen Gasmesser für chemische
Analysen beschreibt J. Bonny in Stolberg (* D.
R. P. Kl. 12 Nr. 12360 vom 1. Juni 1880). Das Meſsgefäſs A (Fig. 1 Taf.
32) ist oben und unten verengt und enthält im Innern den Heber B. Der kurze Schenkel a
des letzteren reicht mit seiner Erweiterung bis auf den Boden, die Biegung c bis in die obere Verengerung von A. Der längere Schenkel b
geht durch den Boden des Gefäſses und mündet unter Wasser in einem Gefäſs C mit gleich bleibender Wasserhöhe. Das Rohr e steht mit der Wasserleitung in Verbindung, das Rohr
f mit der Absorptionsflasche D und der als Wasserverschluſs dienenden Flasche E. Tritt nun durch den Hahn F Wasser in das Gefäſs A, so entweicht das in
diesem befindliche Gas durch die Flasche E. Ist das
Wasser bis zur Biegung c des Hebers B gestiegen, so füllt sich dieser mit Wasser, welches
unten durch b ausflieſst. Sobald nun durch F weniger Wasser eintritt, als durch b ausflieſst, so sinkt dasselbe in A und es wird durch D Gas
angesaugt. Ist dasselbe bis unter den kurzen Schenkel a
gesunken, so. hört die Heberwirkung sofort auf. Sobald der Heber abgelaufen ist,
füllt sich das Gefäſs A von neuem mit Wasser, welches
das Gas durch f und E
hinausdrängt. Damit füllt sich auch der Heber B und
beginnt wieder zu saugen. Die jedesmal durchgesaugte Gasmenge ist gleich dem Volumen
von A zwischen dem höchsten und niedrigsten
Wasserstande. Das Gefäſs A hängt an einer Spiralfeder
G. Beim Entleeren des Gefäſses A zieht diese sich zusammen und bewirkt dadurch, daſs
der Höhenunterschied zwischen A und C derselbe bleibt, so daſs das Gas immer gleichmäſsig
angesaugt wird. Gleichzeitig ist das Gefäſs A mit einem
Hubzähler H verbunden, an welchem die Zahl der
Füllungen abgelesen werden kann.
Der Zugmesser von Tomson in Stolberg besteht aus einem scheibenförmigen
Kästchen von 12cm Durchmesser und 1cm Höhe, welches mit 50cc Alkohol oder Erdöl gefüllt wird. Das
eingetheilte Rohr dreht sich um die durchbohrte Achse m
(Fig. 2 Taf. 32), welche zugleich als Verbindung mit dem Kasten dient. Die Neigung der Röhre
wird durch eine Libelle n geregelt, deren Winkel mit
der Röhre durch eine Schraube v festgestellt werden
kann. Mit einem Zweiwegehahn r kann man abwechselnd den
Kasten mit der atmosphärischen Luft und mit dem Gase, dessen Druck bezieh. Zug zu
messen ist, in Verbindung setzen.
Zur Untersuchung der Rauchgase
verwendet E. Tomson den in Fig. 3 Taf.
32 abgebildeten Apparat, welcher sich namentlich dadurch von anderen (vgl. 1880 237
* 387) unterscheidet, daſs der Hahn A fortwährend durch
den senkrechten Weg in Verbindung mit dem Meſsrohr m
und abwechselnd mit der Gasquelle bei g und mit den
drei Absorptionsgefäſsen a, b und c in Verbindung gesetzt wird. Das Meſsrohr nimmt 10cc Gas auf und ist im schmalen unteren Theile in
0,001 getheilt. Die Flasche d dient zur Herstellung des
richtigen Flüssigkeitstandes in den Röhren, während ein etwas Watte enthaltendes
Kugelrohr g in der Gaszuführung den Staub zurückhalten
soll. Als Untergestell für den Apparat dient die Büchse, in welche der Apparat
eingeschoben und verschlossen wird. Die Füſse können aus der Büchse losgeschraubt
und jeder für sich in zwei Theile zerlegt werden.Der Apparat kostet bei C. Heinz in Aachen 80
M.
Der Apparat zur Bestimmung der
Kohlensäure in Saturationsgasen von A. Kukla
besteht nach der Zeitschrift für Zuckerindustrie in
Böhmen, 1880 Bd. 4 S. 378 aus zwei gleich weiten Röhren M und N (Fig. 4 Taf.
32), von denen die Röhre M in 50cc getheilt und oben mit einem Glashahn K versehen ist. Beide Röhren sind durch die mit dem
Quetschhahn s absperrbare Kautschukröhre T verbunden. Die Flasche L
ist mit einer concentrirten Kochsalzlösung, die Flasche l mit starker Kalilauge gefüllt. Vor Anstellung des Versuches wird aus dem
Gefäſse l die Lauge in die Röhre N bis zu ⅔ der Höhe hinaufgedrückt, worauf nach
Oeffnung des Hahnes K auch die Röhre M in gleicher Weise mit der Kochsalzlösung gefüllt
wird. Dabei ist das Rohr T mit dem Quetschhahn s geschlossen. Hierauf wird mittels des Kautschukrohres
u die Röhre M mit dem
das Saturationsgas enthaltenden Gefäſs in Verbindung gesetzt und die Quetschhähne
a und e geöffnet, so
daſs das eintretende Gas die Kochsalzlösung in die Flasche L drückt. Wenn die ganze Röhre M mit dem Gas
gefüllt ist, wird der Hahn a geschlossen und man läſst
bei geöffnetem Hahn K einige Augenblicke das Gas
hindurchstreichen, um die letzten Reste von Luft zu entfernen. Hierauf werden e und K geschlossen und
s geöffnet. Die Lauge dringt allmählich in die
Röhre M ein. Zur Beschleunigung der Absorption kann die
Röhre M bei r von dem Arm
R abgeschraubt und wiederholt schräg gehalten
werden. Wenn die Lauge in dem Rohre M nicht mehr
steigt, wird es wieder in die ursprüngliche Stellung gebracht und durch Ablassen der
Lauge aus N mittels des Hahnes c die Flüssigkeit in beiden Röhren auf gleiche Höhe gebracht. An der Röhre
M liest man dann die absorbirte Kohlensäuremenge
ab.
Zur Bestimmung des specifischen Gewichtes
von Gasen läſst M. Th. Edelmann (Carl's Repertorium, 1881 S. 261) Gassäulen von gleicher
Höhe auf eine dünne vollkommen elastische Membran drücken und nimmt die Gröſse der
Durchbiegungen, welche durch solche Belastungen der Membran bewirkt wird, als
Maſsstab der Gewichte der Gassäulen. Ansicht und Schnitt des Apparates (Fig.
5 und 6 Taf. 32)
zeigen, daſs am Apparate eine groſse runde, jedoch niedrige Metalltrommel M angebracht ist, deren obere Decke wie bei den
Dosen-Aneroïdbarometern aus einer dünnen, mit concentrischen Wellen versehenen,
vollkommen elastischen Metallscheibe gebildet ist. Diese manometrische Büchse M hängt, am Rande erfaſst, unterhalb eines schweren
guſseisernen Dreifuſses A. Der Rohransatz e ist meist geschlossen, der an f angesetzte Schlauch dient jedoch zur Zuleitung des zu messenden Druckes
auf die Unterseite der Membran, auf deren Oberseite jederzeit der Luftdruck lastet.
Bei der äuſserst geringen Dicke dieser Membran (0mm,1) und bei dem groſsen Durchmesser der Dose (30cm) ist es erklärlich, daſs schon sehr kleine
Druckunterschiede sich bereits als merkliche Durchbiegungen der Membran bemerklich
machen. Diese Bewegungen werden aber noch durch einen Fühlhebel F auſserordentlich vergröſsert, welcher mit einer
Stahlspitze c (Fig. 6)
unter Vermittlung der Spindel o auf der Membran
aufsitzt und hierdurch an allen Bewegungen des Mittelpunktes derselben theilnehmen
muſs, während zwei andere hinter einander liegende Stahlspitzen b, welche gegen feste Theile des Apparates anliegen,
die Stellung der Achse, um welche sich der Fühlhebel dreht, bestimmen. Die Flächen,
auf welchen die drei glasharten Stahlspitzen ruhen, sind mit Steineinlagen wie bei
feinen Wagen versehen.
Ein wesentlicher Bestandtheil dieses Fühlhebels ist ein totalreflectirendes
Glasprisma P, welches alle Drehungen desselben
mitmacht. Mittels eines am Fuſse des Apparates augebrachten Fernrohres D (mit Fadenkreuz) sieht man nach einer etwa 2m entfernten, in Millimeter getheilten Scale. Der
Sehstrahl s dringt jedoch auf seinem Wege durch das
totalreflectirende Prisma, an dessen Hypotenusenfläche sich das Bild der Scale
spiegelt. Dreht sich nun der Fühlhebel sammt dem Prisma um irgend einen
Winkelbetrag, so dreht sich der Sehstrahl wegen der Spiegelung um das doppelte und
führt deshalb andere Scalentheile unter das Fadenkreuz des Fernrohres. Man kann mit
vollkommener Sicherheit Zehntelmillimeter an der Scale ablesen und ein Ausschlag von
0mm,1 bedeutet eine Druckdifferenz von weniger
als einer Millionstel Atmosphäre, oder die Scale müſste unter den vorliegenden Verhältnissen 75m lang sein, um die Druckdifferenz von einer
Atmosphäre ablesen zu lassen.
Um nun das specifische Gewicht von Gasen zu bestimmen, verbindet man eine 2 bis 4mm weite, etwa 2m hohe, senkrechte Glasröhre R (Fig.
5), welche oben etwa auf die Länge von 10cm horizontal umgebogen ist, mit dem Pneumatometer bei f und mit dem Gasentwicklungsapparat oder Behälter bei
g, wobei nur zu beachten ist, daſs f, g, h, i und k ungefähr
in dieselbe Horizontalebene verlegt werden sollen. Zuerst schlieſst man die Hähne
k und h und läſst
durch i das zu untersuchende Gas in die Steigröhre R eintreten und dieselbe füllen. Hierauf schlieſst man
i und öffnet h; der
Gewichtsunterschied zwischen der 2m hohen Gassäule
und einer ebenso hohen Luftsäule wird jetzt seine Wirkung auf die Membran äuſsern.
Nimmt nun der Ausschlag der Scale nicht mehr zu, so werden die genannten Operationen
in der angegebenen Weise einige Male wiederholt, bis der unverändert bleibende Stand
der Scale beweist, daſs nunmehr vollkommen reines Gas sich in der Steigröhre
befindet. Jetzt liest man den Scalenstand im Fernrohre ab, öffnet dann den Hahn k, worauf die Scale in ihre Anfangslage für eine
unbelastete Membran zurücksinkt. Auch dieser Stand wird abgelesen und die
Unterschiede je zweier solcher Ablesungen stellen Bestimmungszahlen für die
specifischen Gewichte der Gase vor. Zur Aichung des Instrumentes bedient man sich
der Messung des Ausschlages für ein Gas von bekanntem specifischen Gewichte, wozu
sich besonders Wasserstoff und Kohlensäure eignen.
Als Beispiel diene die Bestimmung des specifischen Gewichtes g von Leuchtgas. Es wurden abgelesen:
Wasserstoff:
Ausschlag
400,1,
Ruhelage
200,2,
Differenz
a = 199,2
Leuchtgas:
„
309,1,
„
202,8,
„
c = 106,3.
Wenn ς1 = 0,06927 das
specifische Gewicht des Wasserstoffes ist, dann berechnet sich mittels der Formel
\varsigma=1+\frac{(\varsigma_1+1)\,c}{a} das specifische
Gewicht des untersuchten Leuchtgases ς = 0,5051.
Zur Bestimmung der
Verbrennnungswärme der flüchtigen Kohlenwasserstoffe, des Methyläthers u.
dgl. (vgl. S. 145 d. Bd.) verwendet Berthelot nach den
Comptes rendus, 1880 Bd. 91 S. 188 eine 218cc fassende Bombe A
(Fig. 7 Taf. 32) mit abschraubbarem Deckel B,
beide aus 2mm,5 starkem Stahlblech und innen mit
22g Gold galvanisch überzogen. Bei i ist mittels eines Elfenbeineinsatzes ein Platindraht
f eingeführt, um mit Hilfe desselben einen
elektrischen Funken durch das Gasgemisch schlagen zu lassen. Die Bombe wird mittels
der mit dem Ansatz v verbundenen Quecksilberluftpumpe
entleert, das Gasgemisch tritt durch die Bohrung der Schraube C bei c und e ein, worauf der Apparat durch Senken der Schraube bei
e geschlossen wird. Für die Untersuchung saurer
Gase wird die Bombe innen mit Platin ausgekleidet.
W. Louguinine (Annales de
Chimie et de Physique, 1880 Bd. 20 S. 558) verwendete zu seinen in D. p. J. S. 145 d. Bd. besprochenen Versuchen ein
kleines gläsernes Verbrennungsgefäſs v (Fig. 8 Taf.
32), über dessen Asbestdocht bei e ein dünner
Platindraht die beiden dicken Platindrähte p verbindet,
welche mittels der Hartgummiplatte c das
Verbrennungsgefäſs tragen. Um die zu untersuchende Flüssigkeit zu entzünden, läſst
man einen Strom durch die beiden im Stopfen o des
Calorimeters isolirt befestigten starken Platindrähte gehen, so daſs der feine Draht
bei e glühend wird.
Eine Anzahl aus Draht geflochtener
oder gebogener Laboratoriumsapparate beschreibt G. M. Hopkins im Scientific
American, 1880 Bd. 43 S. 354. Hiervon zeigen auf Taf. 32 Fig. 9 einen
Quetschhahn zum Ueberschieben, Fig. 10
einen solchen mit Regulirschraube, Fig. 11 ein
Gestell für Reagirgläschen und Fig. 12
einen Bunsenbrenner, bei welchem die Luftzufuhr durch
Zusammenziehen oder Ausdehnen der Drahtspirale geregelt wird.
Einen tubulirten Tiegel zur
Bestimmung des Kohlenstoffes in Eisen u. dgl. empfiehlt F.
A. Gooch in der Chemical News, 1880 Bd. 42 S.
326. Der cylindrische, etwa 9cm hohe Platintiegel
A (Fig. 13
Taf. 32) ist oben schwach zusammengezogen und mit einer Rinne a versehen, in welche der in ein 5cm langes Platinrohr auslaufende Helm paſst. An
dieses Rohr schlieſst sich bei b ein ⊢-Rohr, welches
bei d mit einem Glasrohr verbunden ist, von dem aus ein
enges Platinrohr c bis in den Tiegel A hineinragt. Bei Ausführung einer Bestimmung bringt
man die zu untersuchende Probe in den Tiegel, setzt den Helm auf und dichtet die
Rinne a durch Einschmelzen von metaphosphorsaurem oder
wolframsaurem Natrium. Nun erhitzt man vorsichtig den Boden des Tiegels, leitet von
d aus durch das Rohr c
Sauerstoff ein und läſst die Verbrennungsproducte in eine passende Vorlage e treten.
Das Löthrohrgebläse von Th. C. Stevens in Galesbury (Amerikanisches Patent Nr.
233 915) wird mit carburirter Luft gespeist. Die Luft wird mittels eines kleinen
Gebläses durch das Rohr a (Fig. 14
Taf. 32) in den Behälter A geblasen, dessen poröse
Füllung B mit Petroleumäther getränkt ist. Die
carburirte Luft geht durch das Rohr c zum Brenner e und in die um die Achse r drehbare Löthrohrspitze f, wodurch eine
sehr heiſse Flamme erzielt werden soll.
Gasbrenner. Wie bereits (1880 238 360) erwähnt, läſst
A. Terquem (D. R. P. Anmeldung Nr. 34076) die Luft
beim Bunsen'schen Brenner von unten zutreten. Auf einem eisernen Fuſse ist das
messingene Gaszuleitungsrohr mit der Ausströmungsspitze c (Fig. 15
Taf. 32) befestigt, über welcher das Brennerrohr a
mittels der Schraube f an den beiden Tragsäulen d auf und nieder bewegt werden kann. Die beiden sich
kreuzenden Metallblättchen stehen etwas über der Brennerröhre hervor und theilen, wie bereits
früher bemerkt, die Oeffnung in vier gleiche Theile. Soll die Lampe in Gebrauch
genommen werden, so schiebe man das Brennerrohr a dicht
auf das Gaszuleitungsstück, zünde das ausströmende Gas an und ziehe das Rohr wieder
in die Höhe, bis an der Basis der Flamme vier kleine Kegel von blaugrüner Farbe
entstehen. Entzündet man das Gas, ohne das Brennerrohr niederzuschieben, so tritt
leicht ein Zurückschlagen der Flamme ein.
Zur Herstellung von monochromatischem Licht wird auf einer hohen Messingsäule das
Gaszuleitungsstück durch die Schraube f (Fig.
16 Taf. 32) gehalten und das Brennerrohr a
durch die Schraube g eingestellt. An der Säule läſst
sich ferner ein sechsarmiger Stern D verstellen, in
dessen federnden Arme sich Eisendrähte von verschiedener Form einschieben lassen.
Der Schornstein e soll die Flamme ruhiger machen und
den oberen Theil derselben verdecken. Die in dem Sterne befestigten Eisendrähte
werden zur Erzeugung von gelbem Lichte vorbereitet, indem man die Drahtösen erst in
Salzwasser und dann in geschmolzenes und gepulvertes Kochsalz taucht. Hierauf
schiebt man die Drähte wieder in die Arme des Sternes und erwärmt das Salz
vorsichtig über kleiner Flamme, bis es zum Schmelzen kommt. Man hebt nun das
Brennerrohr a so weit, bis die kleinen Kegel an der
Basis der Flamme entstehen, und senkt die Salzperlen, bis diese den Gipfel der Kegel
berühren.Stöhrer und Sohn in Leipzig liefern den Brenner
zu 6 M., die vollständige monochromatische Lampe (Fig.
16) zu 18 AI.
Bei dem mehrflammigen Brenner mit
gleichzeitiger Luft- und Gasregulirung von J.
SchoberZu beziehen von J. Schober in Berlin, S. O.
Adalbertstraſse Nr. 44. geschieht die Regulirung sämmtlicher
Flammen einfach durch eine seitliche Bewegung der Schlauchtülle (Fig. 17
Taf. 32), welche nach der einen Seite hin die Zuströmung für Luft und Gas öffnet,
nach der andern Seite hin dieselbe verschlieſst.
Zur Bestimmung des Siedepunktes
verwendet Br. Pawlewski (Berichte der deutschen chemischen Gesellschaft, 1881 S. 88) ein etwa
100cc fassendes Kölbchen K (Fig. 18
Taf. 32), welches halb mit Glycerin, Schwefelsäure, Anilin oder auch mit Paraffin
gefüllt ist. In seinem Halse befindet sich ein Stopfen mit engem Seitenkanal und
einer mittleren Oeffnung, durch welche ein dünnwandiges, 15 bis 20cm langes und 5 bis 7mm breites Probirglas E mit einer kleinen
Oeffnung o geht. Man bringt in das Probirglas 0,5 bis
1cc,5 der zu untersuchenden Flüssigkeit und
befestigt darüber mittels eines Stöpsels ein Thermometer t. Das Quecksilber im Thermometer steigt beim Erwärmen des Apparates rasch
und bleibt bei einem bestimmten Punkte einige Minuten beständig. Dieser Punkt ist
eben der gesuchte Siedepunkt. Auf diesem Punkt bleibt das Quecksilber so lange, als
im Probirglase sich noch
Spuren der Flüssigkeit befinden, obgleich die Temperatur des umgebenden Glycerins 20
bis 40° höher ist als der Siedepunkt der zu untersuchenden Flüssigkeit.
Aleuroskop von H. Sellnick
in Leipzig (* D. R. P. Kl. 42 Nr. 11966 vom 1. Juni 1880). In einem mit Oel
gefüllten, durch eine Spiritusflamme heizbaren Kessel a, mit dem darauf schlieſsenden Deckel durch einen Gummiring verbunden, wird
ein Reagensglas n eingesetzt, welches beim Erhitzen des
Oeles den Backraum bildet, während ein zweites Reagensglas c den zu backenden Kleber aufnimmt. Dasselbe wird durch einen umgelegten
Gummiring vor directer Berührung mit dem gröſseren Glas n geschützt, lose eingesetzt, so daſs es leicht zur augenblicklichen
Beobachtung des Vorganges herausgenommen werden kann. Vergleichende Versuche
erfordern die Anwendung solcher gläserner Backformen von gleichem Durchmesser; auch
können mehrere solcher Apparate in einem gemeinschaftlichen Kessel gefaſst werden.
Die Ausdehnung kann durch ein auſserhalb anzuhaltendes Maſs gemessen werden.
Zum Nachweis des Quecksilbers in
organischen Gemengen wird nach E. Ludwig (Medicinische Jahrbücher, 1880 Sonderabdruck) die durch
Behandlung der Massen mit Salzsäure und chlorsaurem Kalium erhaltene Flüssigkeit mit
Zinkstaub versetzt, dieser nach dem Fällen des Quecksilbers mit Wasser gewaschen und
bei etwa 60° getrocknet. Man schmilzt nun eine 8 bis 10mm weite, schwer schmelzbare Glasröhre an einem Ende zu, füllt nach dem
Erkalten den auf Quecksilber zu prüfenden Zinkstaub a
(Fig. 20 Taf. 32) ein, schiebt darauf in die Röhre einen nicht zu festen
Pfropf aus Asbest x so weit, daſs beim Horizontallegen
der Röhre über dem Zinkstaub ein freier Kanal zum Entweichen der Gase sich bilden
kann, füllt dann grobkörniges Kupferoxyd b nach,
schlieſst dicht an dieses wieder einen Asbestpfropf x
an, auf welchen dann noch eine Schicht von trockenem Zinkstaub c und in ganz geringer Entfernung von diesem wieder ein
Asbestpfropf x folgen. Ist die Röhre in dieser Weise
gefüllt, so wird sie wenige Centimeter neben dem letzten Asbestpfropf zu einer
Capillare e von 1 bis 1mm,5 innerem Durchmesser ausgezogen, an deren Ende man vor der Lampe einen
Wulst f formt zum Anbringen einer Kautschukröhre.
Der vor der Capillare befindliche Zinkstaub hat den Zweck, das aus
dem entgegengesetzten Theile der Röhre beim Erhitzen kommende Wasser zu zerlegen,
also zu verhindern, daſs in der Capillare sich Wassertropfen verdichten; dieser
Zinkstaub muſs vollkommen trocken sein, deshalb vor dem Gebrauche in einem bedeckten
Porzellantiegel ziemlich stark, aber nicht bis zum Schmelzen erhitzt werden. Zuerst
werden die Schichten c und b erhitzt, so daſs das Kupferoxyd rothglühend wird, der Zinkstaub nicht
bis zum beginnenden Schmelzen kommt, und auch die Stelle bei d bis knapp zur Capillare erwärmt, damit sich dort das Quecksilber nicht
condensiren kann, sondern gezwungen wird, sich im Rohre e abzusetzen. Sind c, b und d genügend heiſs geworden, so fängt man an, ganz
behutsam den Quecksilber haltigen Zinkstaub zu erwärmen und steigert die Hitze allmählich und nicht zu
hoch, jedenfalls nicht bis zum Schmelzen des Zinkes. Ist a 10 bis 15 Minuten lang erhitzt, so kann man sicher sein, daſs der
gröſste Theil des vorhandenen Quecksilbers nach e
gewandert ist; man sprengt nun die Röhre bei d ab,
indem man einen Tropfen kalten Wassers auffallen läſst, bringt in den an die
Capillare grenzenden weiten Theil der Röhre bei d, so
lange er noch heiſs ist, einige Körnchen Jod und verbindet f mit einem langsam wirkenden Aspirator, welcher die Joddämpfe durch die
ganze Capillare durchzusaugen hat. Ist alles überschüssige Jod verdampft, so sieht
man, wenn die vorhandene Quecksilbermenge nicht gar zu gering war, an einer oder der
anderen Stelle der Capillare schon fertiges Jodquecksilber als rothen Belag; kann
man einen solchen nicht wahrnehmen, so wird die Capillare über einer sehr kleinen
Flamme erhitzt, indem man sie sehr allmählich in der Richtung von d gegen f durch das
Flämmchen zieht, bis man nahe bei dem Wulste f anlangt;
dort wird dann das etwa vorhandene Jodquecksilber sich concentriren und als rother
Ring sichtbar werden.
Zur Bestimmung des Gesammtstickstoffes
im Harn o. dgl. wird nach E. Ludwig eine
bestimmte Menge desselben, z.B. 5cc, in einem
Schiffchen aus Kupferblech oder Porzellan mit wenigen Tropfen verdünnter
Schwefelsäure auf dem Wasserbad bis auf 2 bis 3 Tropfen verdampft und nach dem
vollständigen Erkalten eine dünne Schicht von pulverförmigem Kupferoxyd in das
Schiffchen gebracht, welches zunächst die Flüssigkeit aufsaugt und sich so mit
derselben innig mischt; nachdem man das Schiffchen weiter mit Kupferoxyd zur Hälfte
angefüllt hat, mischt man dessen Inhalt mit einem Kupferdrahte und spült die an dem
letzteren haftenden Theilchen mit neuem Kupferoxyd nach, mit welchem man das
Schiffchen vollständig anfüllt Das Verbrennungsrohr (Fig. 21
Taf. 32) ist an beiden Enden durch die einfach durchbohrten Kautschukstöpsel b und h verschlossen; das
an einem Ende zugeschmolzene, am anderen Ende verjüngte Glasrohr a ist halb mit kohlensaurem Mangan gefüllt. Bei c in genügender Entfernung von dem Pfropf b befindet sich eine 5 bis 6cm lange Spirale aus Kupferdrahtnetz, welche durch
Erhitzen an der Luft oberflächlich oxydirt ist. Bei d
ist das Schiffchen mit dem eingedampften Harn und Kupferoxyd, bei e eine etwa 15cm
lange Schicht von körnigem Kupferoxyd, bei f eine
Spirale aus Kupferdrahtnetz von 10 bis 12cm Länge,
endlich bei g entweder eine 5 bis 6cm lange Schicht von körnigem Kupferoxyd, oder
eine ebenso lange, durch Erhitzen oberflächlich oxydirte Spirale von
Kupferdrahtnetz. Zur Trennung der Schichten sind Asbestpfropfen x eingeschoben. Der Pfropf h trägt ein Bunsen'sches Kautschukventil, welches durch einen Schlauch mit
dem Apparate zum Aufsammeln und Messen des bei der Verbrennung entwickelten
Stickstoffes (vgl. 1880 237 * 51) in Verbindung steht.
Die Verbrennung wird in folgender Weise ausgeführt: Man legt das
mit dem Kohlensäureentwickler a verbundene Rohr in den
Ofen, ohne zunächst den Apparat zum Ansammeln des Stickstoffes bei h anzufügen, erhitzt das Ende des aus dem Ofen
herausragenden Rohres a direct oder auf einer
Thonunterlage mit einer kleinen Flamme und leitet dadurch die Entwicklung der
Kohlensäure ein. Sobald man annehmen kann, daſs nur noch geringe Mengen von Luft in dem Rohre vorhanden
sind, fügt man den mit Kalilauge gefüllten Meſsapparat an und beginnt nun die
Spirale f und das Kupferoxyd g allmählich zur dunklen Rothglut zu bringen. Inzwischen wird auch, die
Luft aus dem Rohre vollständig verdrängt sein, worüber man in dem Meſsapparat
Aufschluſs erhält; es darf sich in demselben über dem Kali kein unabsorbirbares Gas
mehr ansammeln. Nunmehr wird durch Verkleinern der Flamme unter a der Kohlensäurestrom sehr verlangsamt; ihn ganz zu
unterbrechen, ist nicht rathsam, weil sonst unverbrannte Producte nach a gelangen könnten. Nachdem noch das Kupferoxyd c zu dunkler Rothglut erhitzt worden ist, beginnt man
die einzelnen Abschnitte des Schiffchens d nach
einander zu erhitzen, und wenn dieses auf seiner ganzen Länge so heiſs geworden,
daſs die Entwickelung theerartiger Destillationsproducte nicht mehr zu erwarten ist,
dann wird die Flamme unter a verlöscht und dadurch die
Kohlensäureentwicklung zunächst unterbrochen. Sobald man aus dem Bunsen'schen Ventil
keine Gasblasen mehr austreten sieht, ist die Verbrennung beendet und man kann daran
gehen, durch neuerliches Erhitzen von a an den Stellen,
wo sich noch unzersetztes kohlensaures Mangan befindet, Kohlensäure zu entwickeln
und durch diese die Verbrennungsgase in den Meſsapparat zu drängen.
Nach Beendigung der Verbrennung werden die Flammen des
Verbrennungsofens ausgelöscht, man mäſsigt den Kohlensäurestrom in a, damit er so lange aushalte, bis das Rohr so weit
erkaltet ist, daſs die Spirale f sich nicht oxydiren
kann, entfernt dieselbe dann aus dem Verbrennungsrohre und bewahrt sie als ganz
geeignet für eine zweite Verbrennung auf. Wenn man das Rohr nun erhitzt und bei b aus einem Gasometer Luft durchleitet, so wird alles
reducirte Kupfer oxydirt und das Rohr dadurch für eine weitere Operation in Stand
gesetzt.
Wo viel Stickstoffbestimmungen zu machen sind, empfiehlt E. Ludwig die Anwendung von flüssiger Kohlensäure. An
die Stelle des Rohres a wird dann die etwa 400g flüssige Kohlensäure fassende eiserne Flasche
mit entsprechendem Regulirhahn gesetzt.
Um in offenen Gefäſsen Stoffe mit
flüchtigen Lösungsmitteln zu behandeln, verwendet A, Wynter Blyth (Journal of the Chemical
Society, 1880 S. 140) ein guſseisernes Gefäſs A (Fig. 22
Taf. 32), welches in der Mitte die Schale mit der betreffenden Substanz aufnimmt.
Die Glocke B wird bei a
durch eingegossenes Quecksilber abgeschlossen, während der Hals der Glocke mit einem
Kühler verbunden wird.
Der Apparat zum Auslaugen von
Zucker aus Rüben und anderen Zucker haltigen Rohstoffen (vgl. 1879 234 *
128) ist von C. Scheibler in Berlin (* D. R. P. Kl. 89
Nr. 9481 vom 13. September 1879) jetzt dahin geändert, daſs die auszulaugenden
Stoffe in das unten mit einer filtrirenden Schicht a
(Fig. 23 Taf. 32) geschlossene Rohr b kommen,
welches in dem unten geschlossenen Rohr c steht. Die
entwickelten Dämpfe treten durch kleine Oeffnungen o in
das Mittelrohr b und den aufgesetzten Kühler, die
gebildete Lösung flieſst aus den Oeffnungen e des
Rohres c nach unten und gelangt bei n in die Kochflasche zurück.
Nach Versuchen von B. Tollens (Zeitschrift des deutschen Vereines für Rübenzucker, 1880 S. 483 und 613)
arbeitet dieser Apparat regelmäſsig und gut. Die mit diesem Verfahren erzielten
Resultate sind stets niedriger als diejenigen der Saftpolarisation ausgefallen, und
zwar um 0,4 bis 0,9 Proc. wenn letztere nach 100 : 95 auf die Rübe berechnet waren.
In dem Bleiessigfiltrat der Saftpolarisation ist rechts drehende Substanz, welche nicht
Zucker ist, vorhanden. Ebenso in dem mit Alkohol extrahirten „Rohmark“. Die
Scheibler'sche Extraction lieferte Zahlen, welche
den Resultaten der bei 50 bis 60° ausgeführten Alkoholdiffusion von Rübenbrei sehr
nahe liegen. Die gleichzeitige Extraction von Brei und Preſsrückstand hat Zahlen
gegeben, welche nicht mit den Resultaten der Saftpolarisation und nicht mit der
Annahme von 5 Proc. Colloïdwasser in der Rübe stimmen, welche jedoch am einfachsten
sich erklären lassen, wenn man die Resultate der Scheibler'schen Extraction als richtig annimmt. Da so die theoretischen
Bedenken gegen Scheibler's Verfahren geschwunden sind
und da die Saftpolarisation nicht richtige Zahlen liefert, so liegen die Scheibler'schen Resultate jedenfalls der Wahrheit sehr
nahe.
C. Bögel (Organ des Vereines
für Rübenzuckerindustrie der ö.-u. Monarchie,
1880 S. 39) verwendet zur Bestimmung des Zuckers im Rohzucker mittels Glycerin ein
aus Metall oder Glas gefertigtes Gefäſs A (Fig.
24 Taf. 32), welches ungefähr 3 bis 4cm
vom Boden einen Tubusansatz hat mit kleinem Messinghahn a, welcher durch die Röhre b mit dem Rohre
g des Gefäſses C
verbunden wird. Das Gefäſs B ist in gleicher Weise
durch das Rohr e mit g
verbunden. Das Gefäſs C ist ein Metallkessel, in
welchem der Mittelcylinder D so tief eingeschoben
werden kann, bis er auf dem Ringansatz n feststeht. Der
Metallkessel hat unten einen Siebboden, erhält oben einen Siebboden eingelegt und
kann dann mittels eines aufschraubbaren siebartig durchlöcherten Deckels
verschlossen werden. Auf dem Deckel des Gefäſses D
befindet sich der Hahn k, welcher durch Gummirohr i mit einem Aspirator oder einer Luftpumpe verbunden
werden kann. Ein Hahn o ist durch Rohr p mit der zweihalsigen Woulf'schen Flasche W verbunden, von welcher der Schlauch q ebenfalls zu einem Aspirator oder einer Luftpumpe
führt.
Um nun mit diesem Apparat Rohzucker zu untersuchen, wird derselbe
so aufgestellt, daſs das Gefäſs C mindestens 30cm tiefer steht als die Sohle von A und B; dann werden
sämmtliche Hähne geschlossen und blos q mit dem
Aspirator oder der Luftpumpe verbunden, ohne aber dieselbe in Thätigkeit zu setzen.
Hierauf füllt man Gefäſs A mit reinem Glycerin, welches
so viel wie möglich wasserfrei und auſserdem mit weiſsem Raffinadzucker gesättigt
ist. Die Sättigung des Glycerins mit Zucker erreicht man am besten, indem man das
Glycerin auf ungefähr 500 erwärmt und nun nicht zu fein gestoſsenen Zucker zusetzt.
Man hält das Glycerin ungefähr ½ Stunde bei der Temperatur von 500 und rührt
wiederholt um; hierauf läſst man die Masse langsam bis auf ungefähr 18° abkühlen und
beobachtet dabei, daſs das Glycerin während des Abkühlens nicht gestört wird. Nach
etwa 6 bis 8 Stunden kühlt man rasch das Glycerin auf 0°, noch besser auf – 2° ab,
läſst ungefähr 1 Stunde stehen und gieſst das jetzt gesättigte Glycerin ab. Hat man
zu dieser Arbeit das Gefäſs A benutzt, so braucht man
das Glycerin nicht abzugieſsen.
Man nimmt nun den Einsatz D,
ermittelt von demselben die Tara, füllt denselben ungefähr mit 60 bis 100g gut getrocknetem Rohzucker, verschlieſst D mit seinem Deckel und wiegt. Hierauf setzt man D in C ein, verschlieſst
C
ebenfalls luftdicht und
öffnet nun die Hähne a und c, so daſs das mit Zucker gesättigte Glycerin den Zucker im Siebe D von unten nach oben durchdringt. Man öffnet Hahn o und setzt den Aspirator, welcher mit q verbunden ist, in Thätigkeit, so daſs das Glycerin
von A, nachdem es den Rohzucker in D durchdrungen, nach W
gelangt und hierbei sämmtliche Nichtzuckerbestandtheile löst; dies setzt man einige
Zeit fort, bis der Zucker vollständig rein ist. Inzwischen füllt man Gefäſs B mit absolutem Alkohol, welcher nicht mit Zucker
gesättigt wird, sperrt die Hähne a und c ab, öffnet dafür d und
f, so daſs der Alkohol von B nach C geht und in D den gereinigten Zucker durchdringt. Es ist angezeigt, den Hahn o so weit zu schlieſsen, daſs das Gemisch von Glycerin
und Alkohol nur tropfenweise nach W gelangt, wodurch
die Entfernung des Glycerins am schnellsten erreicht wird, weil der Alkohol Zeit
hat, das Glycerin aufzunehmen. Ist nun anzunehmen, daſs alles Glycerin entfernt ist,
so schlieſst man die Hähne d, f und o und öffnet die Hähne h
und k, so daſs der noch in C befindliche Alkohol abläuft. Hierauf verbindet man i mit dem Aspirator und h
mit einem Apparat zur Erzeugung von trockener, warmer Luft und saugt durch die
Zuckerschicht in D warme trockene oder blos trockene
Luft; in kurzer Zeit ist aller Alkohol verflüchtigt. Man öffnet nun C, nimmt D verschlossen
heraus und wiegt.
Der Unterschied zwischen dem Gewicht von Rohzucker und dem
ermittelten Gewicht des reinen trockenen Zuckers gibt den Gehalt des Rohzuckers an
Nichtzucker an. Da unlösliche Verunreinigungen, Sand u. dgl. nicht in das Glycerin
übergehen, kann man diese direct durch Auflösung des weiſsen Zuckers bestimmen; man
nehme hierzu keinen Alkohol, welcher mit Zucker gesättigt ist, da dann aus dem mit
Zucker gesättigten Glycerin Zucker abgeschieden wird, daher ein Zuviel von Zucker
erhalten würde.
J. Salleron (Comptes
rendus, 1880 Bd. 91 S. 690) berichtet über die Veränderungen der Glasmasse von Aräometern und Thermometern in heiſsen
Flüssigkeiten. Aräometer erleiden nicht selten innerhalb weniger Tage
Gewichtsverminderungen von 0g,5, wodurch
Messungsfehler von 6 bis 8° B. bedingt werden. Bei anderen Aräometern werden die
cylindrischen Behälter aufgeblasen, so daſs derartige Apparate, wie auch die
Thermometer häufig controlirt werden sollten.
Um das Zerspringen der Glasaräometer für
heiſse Flüssigkeiten zu verhüten, schlagen J. C.
Primavesi und Sohn in Magdeburg (* D. R. P. Kl. 42 Nr. 11960 vom 21. April
1880) vor, den unteren Theil derselben galvanisch mit einer 0mm,5 dicken Metallschicht zu überziehen.
Das Margarimeter von Leune und Harburg besteht nach der Milchzeitung, 1881 S. 122 aus einem kleinen Kessel a (Fig. 25
Taf. 32), in welchem etwa 500c° Wasser zum Sieden
erhitzt werden, während das mit Mantel b umgebene
Metallgefäſs c mit der vorher geschmolzenen Butter
gefüllt wird. Der Nullpunkt des eingetauchten Aräometers e soll reine Butter, jeder folgende Grad 10 Proc. fremdes Fett anzeigen.
(Vgl. 1880 235 148. 1881 239 150.)
Tafeln