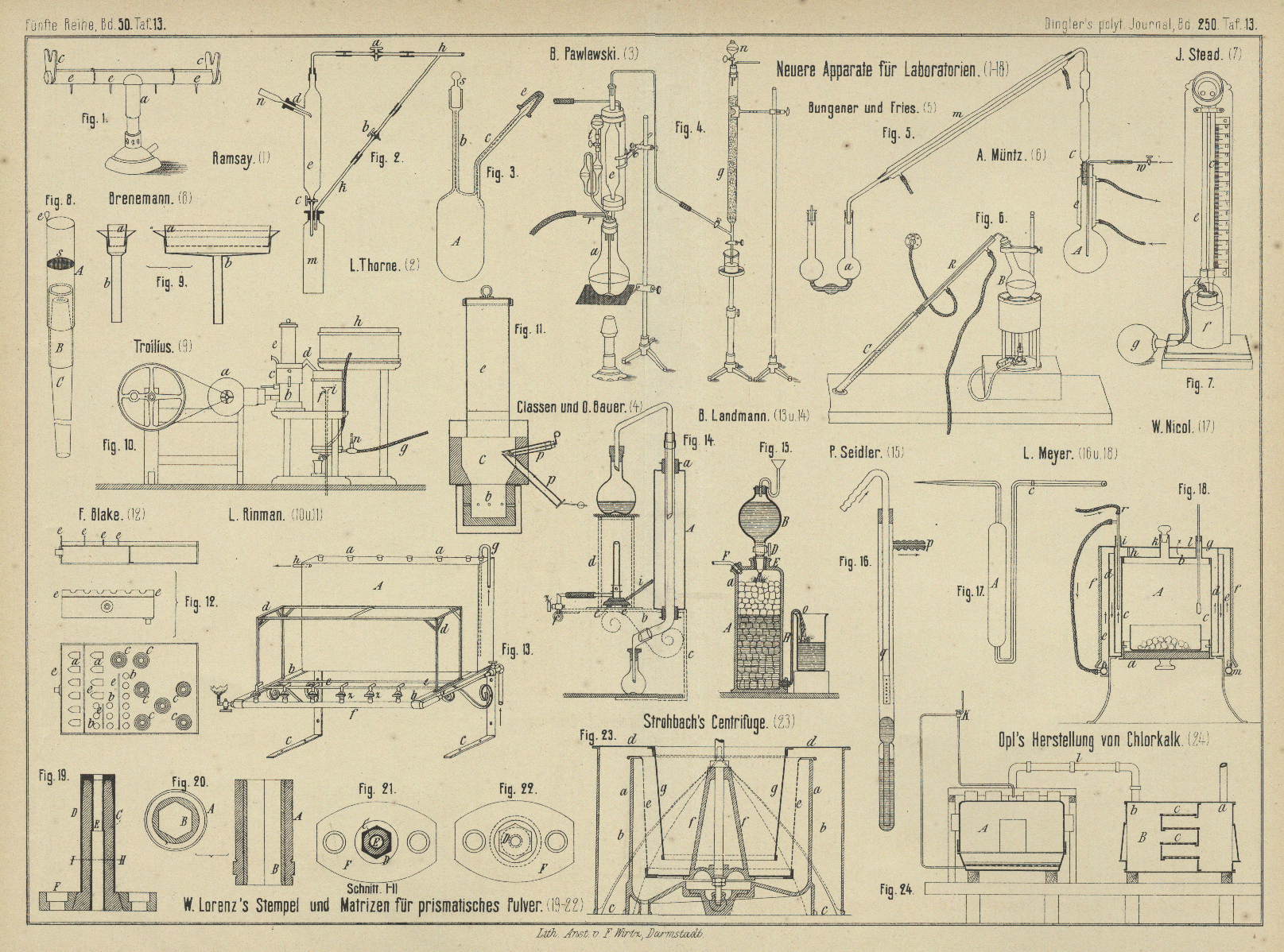
| Titel: | Neuere Apparate für Laboratorien. |
| Fundstelle: | Band 250, Jahrgang 1883, S. 160 |
| Download: | XML |
Neuere Apparate für Laboratorien.
Mit Abbildungen auf Tafel 13.
Neuere Apparate für Laboratorien.
Einen breitflammigen Bunsen'schen Brenner stellt W. Bamsay (Chemical News, 1883 Bd. 48 S. 2) dadurch
her, daſs er auf einen gewöhnlichen Bunsen sehen
Brenner ein mit Ansatz a (Fig. 1 Taf.
13) versehenes Messingrohr setzt, welches oben der ganzen Länge nach geschlitzt, an
beiden Enden aber geschlossen ist. Zur Regulirung der Flamme sind mit kleinen
Griffen versehene, oben ebenfalls geschlitzte Hülsen e
übergeschoben, durch deren Drehung der Hauptspalt mehr oder weniger abgestellt
werden kann. Die zu erhitzenden Glasrohre werden von kleinen Ständern c getragen.
L. T. Thorne beschreibt in den Berichten der deutschen chemischen Gesellschaft, 1883 S. 1327 einen Apparat zur fractionirten Destillation unter vermindertem
Drucke. Das 10 bis 12cm lange Rohr e (Fig. 2 Taf.
13) ist unten mit einem Hahne c verschlossen und oben
ein Ansatz d angeschmolzen, um die Spitze des Kühlers
n einzulassen. Das Knierohr h verbindet den Hahn a mit dem Dreiweghahne
b, während der dritte Schenkel zur Luftpumpe führt.
Um namentlich bei Anwendung einer Wasserstrahlluftpumpe Unregelmäſsigkeiten im
Vacuum auszugleichen, empfiehlt sich die Einschaltung einer Flasche von etwa 3l Inhalt zwischen der Pumpe und dem Rohre h. Beim Gebrauche wird Hahn a geöffnet, b so gedreht, daſs die Rohre h und k in Verbindung
stehen, die Pumpe in Thätigkeit gesetzt und mit der Destillation begonnen. Sobald
die erste Fraction übergegangen oder das Rohr e gefüllt
ist, wird der Hahn c geöffnet, wobei das Destillat
gleich in das Gefäſs m herunterflieſst. Sollte das
Destillat etwa dickflüssig sein und nicht leicht flieſsen, so wird der Hahn a zugemacht, wodurch die Pumpe durch b und c mitwirkt; der Hahn
c wird dann wieder geschlossen. Wenn man das Gefäſs
m wechseln will, wird der Dreiwegehahn b so gestellt, daſs m mit
der Luft in Verbindung, h dagegen zugesperrt steht.
Wenn ein neues Gefäſs angepaſst worden ist, wird a für
ein paar Secunden zugemacht und Rohr h mit k wieder durch b in
Zusammenhang gebracht, dann, sobald m geleert, a wieder geöffnet. Auf diesem Wege kann eine beliebige
Anzahl Fractionen
abgenommen werden, ohne auch nur einen Augenblick die Destillation unterbrechen zu
müssen.
Zur Dampfdichtebestimmung empfiehlt B. Pawlewski (Daselbst S. 1293) ein kleines, 20 bis
30cc fassendes Gefäſs A (Fig. 3 Taf.
13), so daſs man nur 0,5 bis 1g Substanz
gebraucht. Das kegelförmige Ende des Rohres c wird mit
einem gut aufgeschliffenen Hütchen e, das Rohr b mit einem Stopfen s
geschlossen. – Fr. Müller in Bonn liefert den Apparat
für 3,50 M.
A. Classen und O. Bauer
empfehlen a. a. O. S. 1061 die Verwendung des
Wasserstoffsuperoxydes in der analytischen Chemie, Während
Schwefelwasserstoff nach Zusatz von Wasserstoffsuperoxyd Schwefel abscheidet: H2O2 + H2S = 2H2O + S, geben
Schwefelammonium oder Schwefelnatrium mit Wasserstoffsuperoxyd bei gewöhnlicher
Temperatur schwefelsaures und unterschwefligsaures Salz, beim Kochen nur Sulfat.
Versetzt man die Lösungen von Schwefelzinn, Schwefelantimon und Schwefelarsen in
Schwefelammonium nach und nach mit Wasserstoffsuperoxyd, so wirkt dasselbe zunächst
oxydirend auf das Schwefelammonium ein und es entstehen vorübergehend Niederschläge
von Zinnsulfid, Schwefelantimon und Schwefelarsen. Durch einen Ueberschuſs des
Reagens und Erwärmen geht nun das Zinnsulfid quantitativ in unlösliches Oxyd, das
Antimon theilweise in unlösliches Oxyd, theilweise in eine lösliche
Antimonverbindung, das Schwefelarsen aber quantitativ in lösliches arsensaures Salz
über. Bei Einwirkung von Wasserstoffsuperoxyd auf Natriumzinnsulfid bleibt je nach
der Menge von Schwefelnatrium, welche zur Bildung des Sulfosalzes angewendet wurde,
entweder die ganze oder die gröſste Menge des Zinnes in Auflösung. Die
Schwefelverbindungen von Arsen, Kupfer, Zink und Thallium werden durch
ammoniakalisches Wasserstoffsuperoxyd ohne Abscheidung von Niederschlägen oxydirt.
Zinnsulfid wird unter Abscheidung von Oxyd und Oxydation des Schwefels zu
Schwefelsäure zersetzt. Schwefeleisen gibt Schwefelsäure und Hydroxyd,
Schwefelmangan dagegen Superoxydhydrat und Oxydhydrat. Beim Erwärmen von
Schwefelkobalt mit ammoniakalischer Wasserstoffsuperoxydlösung wird zuerst lösliches
Kobaltsulfat gebildet, welches beim weiteren Erhitzen theilweise, unter Abscheidung
eines schmutzig braunen Niederschlages, weiter angegriffen wird. Schwefelnickel wird
unter Ausscheidung eines grünen Niederschlages, welcher ebenfalls nicht alles Nickel
enthält, ähnlich wie Schwefelkobalt zersetzt. Die Schwefelmetalle von Silber und
Wismuth werden durch ammoniakalisches Wasserstoffsuperoxyd nicht angegriffen.
Schwefelblei gibt Sulfat.
Die Eigenschaft des Wasserstoffsuperoxydes, in alkalischer Lösung Schwefelwasserstoff
leicht und vollständig zu Schwefelsäure zu oxydiren, kann nun zunächst zur
Bestimmung von Chlor-, Brom- und Jodwasserstoffsäure in Schwefelwasserstoff
enthaltenden Flüssigkeiten benutzt werden. Zu diesem Zwecke versetzt man letztere
mit Natriumcarbonat und
Wasserstoffsuperoxyd, kocht, bis sich keine Sauerstoffbläschen mehr entwickeln, und
fällt in gewöhnlicher Weise mit Silbernitrat und Salpetersäure.
In den durch Wasserstoffsuperoxyd direkt oxydirbaren Schwefelmetallen kann die Menge
des Metalles aus der gebildeten Schwefelsäure berechnet werden. Dieses Verfahren ist
z.B. anwendbar bei den Schwefel Verbindungen von Arsen, Zink, Kupfer und Kobalt
sowie bei Antimontrisulfid, während Antimonpentasulfid durch Wasserstoffsuperoxyd
nur unvollständig oxydirt wird.
Schwefelmetalle, welche sich durch Kochen mit Chlorwasserstoffsäure unter
Entwickelung von Schwefelwasserstoff auflösen, können durch Ueberführung desselben
in Schwefelsäure bestimmt werden. Zu diesem Zwecke ist das Kölbchen a (Fig. 4 Taf.
13), welches zur Aufnahme der zu zersetzenden Schwefelmetalle dient, mit einem
dreifach durchbohrten Stopfen verschlossen. In die eine Durchbohrung reicht das
Abzugsrohr e, in die zweite ein Trichterrohr t und in die dritte ein für Kohlensäuregas bestimmtes
Einleitungsrohr r. Das Abzugsrohr ist mit einem Kühler
umgeben, welcher zur Condensation der Chlorwasserstoffsäure dient, und steht in
Verbindung mit einem zweiten, aufrecht stehenden Glasrohre g, das mit Glasperlen gefüllt ist und in welchem das
Schwefelwasserstoffgas durch beständig herabtropfendes Wasserstoffsuperoxyd oxydirt
wird. Die im Glasrohre sich ansammelnde Flüssigkeit kann durch einen Glashahn
abgelassen werden. Man bringt die Probe in das Kölbchen, läſst durch das
Trichterrohr etwa 50cc verdünnte Salzsäure
zuflieſsen und dann Kohlensäure durch die Flüssigkeit streichen. Gleichzeitig läſst
man aus dem Tropftrichter n Wasserstoffsuperoxyd in
alkalischer Lösung in die Röhre g eintropfen und am
unteren Ende so abflieſsen, daſs die Röhre zu ⅓ mit Flüssigkeit gefüllt bleibt. Den
Inhalt des Kölbchens erhitzt man nun zum Kochen, spült nach 15 bis 20 Minuten die
Absorptionsröhre g mit Wasser, säuert die Flüssigkeit
vorsichtig mit Chlorwasserstoffsäure an, kocht zur Zersetzung des
Wasserstoffsuperoxydes und fällt mit Chlorbarium.
Bezügliche Versuche mit Antimontrisulfid, Antimonpentasulfid (Sb2S5 + 6HCl =
2SbCl3 + 3H2S +
2S), Zinnsulfid, Schwefelcadmium und Schwefeleisen, sowie die Bestimmung des
Schwefels im Roheisen fielen befriedigend aus.
Zur Bestimmung von Schwefligsäure in irgend einem Sulfite wird genau wie bei
Bestimmung von Schwefelwasserstoff in einem Schwefelmetalle verfahren. Enthält das
Sulfit kein Sulfat, so kann die Ueberführung in Schwefelsäure durch direkte
Einwirkung einer alkalischen Lösung von Wasserstoffsuperoxyd geschehen. Im anderen
Falle treibt man die Schwefligsäure durch Kochen mit verdünnter
Chlorwasserstoffsäure aus und oxydirt das Schwefeldioxyd wie Schwefelwasserstoff.
Dieses Verfahren gestattet die Bestimmung von Natriumhyposulfit, Natriumsulfit und Sulfat neben
einander. Man zersetzt die abgewogene Substanz mittels Chlorwasserstoffsäure im
Apparate, bestimmt das Schwefeldioxyd, welches dem Sulfite nebst Hyposulfit
entspricht, filtrirt den im Kölbchen zurückgebliebenen Schwefel auf gewogenem Filter
und fallt im Filtrate die Schwefelsäure als Bariumsulfat. Aus der Menge des
Schwefels ergibt sich die des Hyposulfites bezieh. die des Sulfites.
Die Bestimmung des Stickstoffes nach dem von W. Bettel vorgeschlagenen Verfahren durch Behandeln der
Probe mit geschmolzenem Natron in einer kupfernen Flasche unter Einleiten von
Wasserstoff und Auffangen des gebildeten Ammoniaks in titrirter Schwefelsäure ist
nach H. Bungener und L. Fries
(Zeitschrift für das gesammte Brauwesen, 1883 S. 40) besonders dann zu
empfehlen, wenn es sich um die Analyse solcher Substanzen handelt, welche entweder
flüssig, oder schwer zu pulvern sind. Mit einiger Uebung bekommt man, z.B. mit Bier,
Würze, Gerste und Malz, sehr scharf übereinstimmende Resultate. Die Handhabung des
Apparates ist bequem und es kann eine Analyse in weniger als 2 Stunden ausgeführt
werden. Die Methode ist billiger als die gewöhnliche, da der Apparat für Hunderte
von Bestimmungen gebraucht werden kann und der Gasverbrauch verhältniſsmäſsig gering
ist.
Der Apparat besteht passend aus einer etwa 300cc
fassenden kupfernen Flasche 4 (Fig. 5 Taf.
13), deren gröſste Wanddicke am Boden 3 bis 4mm
beträgt. Der 15cm lange, 3cm weite Hals, welcher wo möglich aus Messing
hergestellt wird, ist mit einem aus Blech angefertigten Kühler e umgeben. Um nun z.B. den Stickstoff in einem Biere zu bestimmen, gieſst man in die Flasche die
concentrirte Lösung von 15 bis 20g kaustischem
Natron und dann 20 bis 25cc Bier, setzt den
Stöpsel auf, verbindet die eine Röhre w mit dem
Wasserstoffapparate, die andere c mit dem
Absorptionsgefäſse a, welches 30cc 1/10-Normal-Schwefelsäure enthält, füllt den Kühlere mit Wasser, erhitzt den
Boden der Flasche vorsichtig und läſst einen langsamen Strom Wasserstoff durch den
Apparat gehen. Der Inhalt der Flasche fängt bald an zu sieden; das Wasser im Kühler
erwärmt sich allmählich und nach einiger Zeit steigt der Dampf durch die Glaswolle
im Rohre c. Ein Theil verflüssigt sich in c und bildet hier eine 3 bis 4cm hohe Schicht siedenden Wassers, durch welche
der übrige Dampf, der Wasserstoff und später die flüchtigen Zersetzungsproducte in
die Vorlage destilliren. Sobald der Inhalt der Flasche zur Trockne eingedampft ist,
macht man die Flamme gröſser, so daſs der Boden der Flasche allmählich bis zur
beginnenden Rothglut erhitzt wird, welche Temperatur man 20 Minuten bis ½ Stunde
unterhält. Dabei muſs man Sorge tragen, daſs der Kühler mit Wasser gefüllt bleibt.
Nach 20 bis 30 Minuten nimmt man die Flamme weg und läſst den Wasserstoffstrom etwas
rascher gehen, damit keine Luftleere in der Flasche entsteht und das Wasser nicht
von c hineingesaugt wird, was eine heftige
Dampfentwickelung zur Folge haben würde. Hat sich die Flasche bis auf 100°
abgekühlt, so unterbricht man den Wasserstoffstrom; das in der Röhre c verflüssigte Wasser flieſst zurück. Man erhitzt
alsdann wieder wie anfangs und wiederholt die ganze Operation; doch braucht man
diesmal die Flasche nach Verdampfung des Wassers nur etwa 10 Minuten zu erhitzen.
Man nimmt nun die Vorlage ab und titrirt den Inhalt auf gewöhnliche Art.
Um den Stickstoff in Gerste und Malz zu bestimmen, bringt man etwa 1g
fein gemahlene Substanz in die Flasche A, setzt 15 bis
20g Natron in etwa 50cc Wasser gelöst hinzu und verfahrt wie beim
Biere. Will man den Kühler m weglassen, so muſs man die
Vorlage a in kaltes Wasser setzen. Die Resultate fallen
bei Gerste und Malz nach diesem Verfahren etwas höher aus als mit Natronkalk,
wahrscheinlich, weil solche schwer fein zu pulvernden Stoffe mit Natronkalk nicht
immer vollständig zersetzt werden.
A. Müntz (Comptes rendus, 1883 Bd. 96 S. 1430) benutzt
zur Bestimmung des Schwefelkohlenstoffes in
Sulfocarbonaten die Löslichkeit des Schwefelkohlenstoffes in Erdöl. Zu
diesem Zwecke bringt Müntz in einen Halbliterkolben B (Fig. 6 Taf.
13) 30cc Sulfocarbonat mit 100cc Wasser und 100cc einer gesättigten Lösung von schwefelsaurem Zink. Das 60cc fassende Meſsrohr C enthält 30cc Erdöl, in welches die
ausgezogene Spitze des vom Kühler R umgebenen
Gasentwickelungsrohres eintaucht. Man erwärmt im Dampf bade, bis auſser dem
Schwefelkohlenstoffe etwa 10 bis 12cc Wasser mit
übergegangen sind, welches sich von der Lösung des Schwefelkohlenstoffes in Erdöl
klar abscheidet, und liest die Volumenzunahme ab unter Berücksichtigung von 0cc,2 als Berichtigung für den noch im ausgezogenen
Rohre zurückgebliebenen Antheil Schwefelkohlenstoff.
Bei einem Versuche betrug z.B. das Volumen des Erdöles 31cc,1, der Flüssigkeit nach beendeter Destillation 49cc,6, des Wassers 13cc,8, somit des Erdöles und Schwefelkohlenstoffes 35,8 und des
Schwefelkohlenstoffes 4cc,7 oder mit Berichtigung
4cc,9, entsprechend 6g,22 für 30cc
Sulfocarbonat oder 14,8 Proc.
Zur colorimetrischen Bestimmung des Kohlenstoffes in Eisen
und Stahl übergieſst man, wie J. Stead im Iron, 1883 Bd. 21 S. 454 mittheilt, 18 der Probe in
einem bedeckten Becherglase mit 12cc Salpetersäure
von 1,2 sp. G. und erwärmt bis zur völligen Lösung auf 100°. Gleichzeitig behandelt
man eine Eisenprobe von bekanntem Kohlenstoffgehalte in derselben Weise und versetzt
beide Lösungen mit 30cc heiſsem Wasser und 13cc Natronlauge von 1,27 sp. G., schüttelt,
verdünnt auf 60cc und filtrirt. Die durch den
Kohlenstoffgehalt des Eisens bewirkte Färbung ist in alkalischer Lösung etwa 2,5 mal
so stark als in saurer.
Zur Vergleichung der Farben wird die Normalfarblösung in eine 125cc fassende Flasche f
(Fig. 7 Taf. 13) gefüllt, durch deren Stopfen das Rohr c bis auf den Boden der Flasche reicht, während das
kürzere Rohr a mit einem Gummiballe g
verbunden ist. Ueber dem verengten Theile des Rohres c
befindet sich ein kleiner glasirter Porzellancylinder und ein gleicher ist am
unteren Ende des Rohres e angebracht, welches bis zu
einer Marke mit der zu vergleichenden Lösung gefüllt wird. Ueber den offenen Enden
beider Röhren ist ein kleiner Spiegel unter einem Winkel von 45° befestigt. Man
treibt nun mittels des Gummiballes die Flüssigkeit im Rohre c so hoch, daſs die Farbstärke beider Lösungen gleich ist. Der
Kohlenstoffgehalt steht dann im umgekehrten Verhältnisse zur Länge der
Flüssigkeitssäulen.
Bezügliche Versuche ergaben, daſs nach dem Lösen der Eisenprobe noch etwa 5 Minuten
erwärmt werden muſs, um den Kohlenstoff völlig zu lösen; ein längeres Erwärmen hat
wenig Einfluſs auf die Farbstärke. Werden statt 12 mehr als 18cc Salpetersäure verwendet, so wird dadurch die
Färbung verringert. Nimmt man weniger als 13cc
Natronlauge, so wird ein Theil des Farbstoffes mit dem Eisenhydrate gefällt. Während
die Färbung der sauren Lösung schon durch Spuren von Salzsäure beeinfluſst wird, ist
dies bei der alkalischen Lösung nicht der Fall. Bei Proben mit nur geringem
Kohlenstoffgehalte ist der Einfluſs des Härtens auf dieses colorimetrische Verfahren
nur gering.
Stead hat ferner gefunden, daſs in den sauren
Stahllösungen ein gelber, dem Kaliumchromate in Farbe ähnlicher und ein
dunkelbraunrother Farbstoff in wechselnden Verhältnissen vorhanden sind (vgl. Chemical News, 1883 Bd. 47 S. 285).
A. Brenemann (American Chemical Society, 1883 Bd. 5 S.
56) verwendet bei der Bestimmung des Kohlenstoffes in
Eisen ein mit Gold ge-löthetes Rohr aus Platinblech A (Fig. 8 Taf.
13), welches mit Hilfe eines Kautschuckschlauches B
fest auf ein kegelförmiges, mit einem Saugapparate verbundenes Rohr C gesetzt wird. Das kleine Platinsieb s wird mit Asbest bedeckt, dann wird filtrirt und das
Rohr A mit dem auf dem Asbeste gesammelten Kohlenstoff
an der Oese e in das Verbrennungsrohr geschoben.
M. Troilius (Jern-Kontorets Annalen, 1882 Heft 8 durch
die Berg- und Hüttenmännische Zeitung, 1883 S. 255)
verwendet zur Bestimmung des Kohlenstoffes in Eisen
entsprechend dem A. Blair'schen Verfahren 3g Roheisen, Spiegeleisen oder Ferromangan, bezieh.
5g Stahl als Pulver oder Bohrspäne, welche mit
einer Lösung von Kupferammoniumchlorid, Cu(NH4)2Cl4, behandelt
werden. Zur Herstellung dieser Flüssigkeit löst man 2k des Salzes in 5l Wasser und setzt
Ammoniak zu, bis die Fällung nicht mehr verschwindet. Hiervon verwendet man für je
1g der Probe 50cc und 50cc zum Auflösen des
ausgefallenen Kupfers, läſst unter Umrühren ¼ Stunde bei gewöhnlicher Temperatur
einwirken und erwärmt dann auf höchstens 50°, bis das Kupfer gelöst ist. Etwa
ausgeschiedenes basisches Eisensalz wird durch einige Tropfen Salzsäure entfernt.
Zum Filtriren verwendet man ein Platinschiffchen a
(Fig. 9 Taf. 13), dessen Siebboden mit Asbest bedeckt ist und das in den
Platintrichter b gesetzt wird, welchen man in dem
Stopfen einer mit Saugapparat verbundenen Flasche befestigt. Der auf dem Asbeste
zurückgebliebene Kohlenstoff wird zuerst mit Salzsäure von 1,2 sp. G., dann mit
Wasser ausgewaschen, bei 100° getrocknet und mit dem Schiffchen in das
Verbrennungsrohr geschoben, um ihn im Sauerstoffstrome zu verbrennen. Blair verwendet hierzu ein 0m,6 langes Platinrohr, welches ein 15cm langes Platinnetz enthält; – ein Glasrohr mit
einer kurzen Schicht von platinirtem Asbest würde wohl dasselbe leisten.
Bei dem Apparate zur Untersuchung der Brennstoffe von
L. Rinman (Berg- und Hüttenmännische Zeitung, 1883
S. 258) wird in den Verbrennungsraum des Generators b
(Fig. 10 und 11 Taf. 13)
Luft durch ein kleines Gebläse a eingetrieben. Die
Erhöhung c ist mit einem Deckel aus Kupferblech
abgeschlossen, auf welchem der Aufgebetrichter oder Cylinder e befestigt ist. Das Gas geht durch das Kupferrohr d zum Condensator f, aus 13 Messingröhren von
37mm Durchmesser und 585mm Länge bestehend, welche mit Wasser aus dem
Behälter h gekühlt werden. Das verbrauchte Wasser läuft
am oberen Theile des Condensators in den Trichter i ab.
Das abgekühlte Gas geht, das Thermometer n streifend,
durch den Kautschukschlauch g in ein anderes Zimmer zu
einem Hahne, durch dessen entsprechende Stellung das Gas abwechselnd in einen der
beiden Gasometer von je 40l Inhalt tritt, um nach
geschehener Messung von hier in einen Schornstein zu entweichen. Zum Messen der
Erzeugungstemperatur des Gases wird durch die Porzellanrohre p eine Platinkugel eingeführt.
Bei der Verbrennung von 12g,5 Holzkohle in der
Minute ergab der Rinman'sche Apparat folgende
Generatorgase:
Gewichtsprocent
Kohlensäure
2,73
2,59
Kohlenoxyd
32,23
32,65
Grubengas
–
0,04
Wasserstoff
0,40
0,39
Stickstoff
64,64
64,33.
Die Erzeugungstemperatur der Gase war 660° und 670°. Hierbei
ist zu bemerken, daſs der Generator mit warmer Luft von etwa 230° gespeist
wurde.
F. C. Blake beschreibt in der Mining and Scientific Press, 1883 Bd. 46 S. 209 ein Wasserbad aus Kupferblech, welches im Laboratorium der
Pennsylvania Lead Company verwendet wird. Die
Löseflaschen für die Gay-Lussac'schen Silberproben
werden so in die Ausschnitte a (Fig. 12
Taf. 13) gelegt, daſs sie mit dem Halse auf den Stützblechen c ruhen, so daſs Verluste durch Spritzen vermieden werden. Die kleineren
Oeffnungen b nehmen die Lösefläschchen für die
Quartationsprobe auf, während die mit über einander greifenden Kupferringen
bedeckten Oeffnungen c zum Erwärmen von Bechergläsern
u. dgl. dienen.
Der Destillationsapparat für Alkoholbestimmungen in
Weinen von B. Landmann (Zeitschrift für analytische
Chemie, 1883 S. 394) ermöglicht bei thunlichster Raumersparniſs mehrere
Destillationen neben einander ausführen zu können. Derselbe besteht aus einem
gemeinschaftlichen 54cm laugen und 30cm hohen Kühlgefäſse A (Fig. 13 und
14 Taf. 13) von Blech, mit den Oeffnungen a
für das Durchführen der Kühlrohre. Das Kühlwasser flieſst von g nach h. Das Kühlgefäſs
steht auf den 23 bis 24cm langen eisernen Trägern
b, an welchen am vorderen Ende das Gasleitungsrohr
f mit sechs 5cm
langen Hähnen z und einer Leuchtflamme angeschraubt
ist. Die Träger sind fest verbunden durch die beiden parallelen Stangen e und durch das eiserne Gestell d, welches 4cm vom Kühlgefäſse
angebracht ist, eine Höhe von 20cm und eine Breite
von 7cm hat. Zum Tragen der Auffanggefäſse dient
ein auf die Träger c 20cm von b entfernt befestigtes Brett. Das
Brett i soll die strahlende Wärme der Flammen von den
Ausfluſsmündungen abhalten.
Der in Fig. 15 Taf. 13 gezeichnete Gasentwickelungsapparat von P. Seidler
(Daselbst S. 529) zur Herstellung von Kohlensäure, Wasserstoff und
Schwefelwasserstoff besteht im Wesentlichen aus dem Entwickelungsgefäſse A, dem Säureballon B und
dem Steigerohre H. Das Entwickelungsgefäſs wird z.B.
für die Darstellung von Kohlensäure etwa bis zur Marke a mit Kalksteinstückchen angefüllt und durch den Stutzen E concentrirte Chlorcalciumlösung hinzu gegeben, bis
diese beginnt, bei o auszutreten. Alsdann setzt man den
mit Salzsäure gefüllten Ballon B auf E und öffnet den Hahn D,
worauf die Säure in dünnem Strahle aus B austritt. Es
bildet sich auf bekannte Weise Chlorcalcium und Kohlensäure. Letztere tritt durch
Rohr F aus, während die Salzsäure langsam von oben nach
unten die Kalksteinfüllung durchflieſst und durch Rohr H als neutrale Chlorcalciumlösung abläuft.
L. Meyer (Berichte der deutschen chemischen
Gesellschaft, 1883 S. 1087) verwendet für Luftbäder einen verbesserten Kemp'schen
Regulator, welchen er ganz aus Glas, nur das untere Ende des Zuleitungsrohres aus
durchlöchertem Platinbleche, herstellen läſst. Um ihn zu füllen, ersetzt man das
Zuleitungsrohr durch ein an beiden Enden ausgezogenes, bis in die Kammer des
Regulators reichendes Glasrohr, verbindet den Seitenansatz p (Fig. 16
Taf. 13) mit der Wasserluftpumpe, kehrt den Regulator um und taucht das Ende des
ausgezogenen Rohres einen Augenblick in die einzubringende Flüssigkeit, dann in
Quecksilber, bis die Kammer nahezu, aber noch nicht ganz gefüllt ist. Alsdann wird
der Apparat aufgerichtet, ein wenig Quecksilber nachgegossen und das Zuleitungsrohr
q eingesetzt. Beim Gebrauche wird letzteres
zunächst in die Höhe gezogen und, sobald das Luftbad der gewünschten Temperatur nahe
kommt, mit seinem unteren Ende bis in das Quecksilber eingeschoben, so daſs die
Gaszufuhr auf ein Minimum beschränkt wird. Durch vorsichtiges Schieben läſst sich
leicht die Stellung finden, in welcher die Spannung des in der Kammer entwickelten Dampfes das
Quecksilber gerade so weit empordrückt, daſs bei der richtigen Temperatur die untere
Oeffnung des Gaszuleitungsrohres eben gesperrt wird. Da das Luftbad sich sehr
langsam abkühlt, aber rasch erwärmt, ist es zweckmäſsig, den Regulator zunächst auf
eine etwas zu niedrige Temperatur einzustellen. Man läſst sich zweckmäſsigerweise
eine gröſsere Zahl solcher Regulatoren blasen, beschickt sie mit Stoffen, deren
Siedpunkte ungefähr 30° aus einander liegen, und hebt sie in einem passenden, dem
für Probircylinder ähnlichen Gestelle der Reihe nach geordnet zum Gebrauche auf.
Geeignete Stoffe sind für Wasserbäder: Chloräthyl, Aether, Schwefelkohlenstoff,
Gemische aus Aether und Alkohol, reiner Alkohol oder Benzol, ferner für Luftbäder:
Wasser, Toluol, Xylol oder Amylalkohol, Cumol oder Terpentinöl, Anilin oder Phenol,
Naphtalin, Diphenyl oder Diphenylmethan, Diphenylamin und allenfalls noch Anthracen.
Reinheit der Stoffe ist nicht erforderlich; die bei gewöhnlicher Temperatur starren
sind sogar im unreinen Zustande bequemer, da sie niedriger schmelzen.
Das in Fig. 18 Taf. 11 ersichtliche Luftbad ist aus 4 Kupferblechcylindern
zusammengesetzt. Der innere Cylinder c umschlieſst den
zu erhitzenden Raum A, welcher unten durch den mit
Bajonetverschluſs einzusetzenden Doppelboden a, oben
durch den Deckel b verschlossen werden kann. Auf 3
Trägern h des letzteren ruht der zweite Deckel g, welcher den Tubus i für
den Regulator r trägt und von zwei Löchern für die
Tuben k und l sowie von in
zwei concentrischen Kreisen stehenden kleinen Löchern für den Durchtritt der
Heizgase durchsetzt wird. Mit diesem Deckel g sind die
beiden Cylinder d und f
fest verbunden, während e mit c unten zusammenhängt und mit ihm von drei Füſsen getragen wird. Der
Apparat läſst sich daher ganz zerlegen. Die Heizung geschieht durch den mit
regulirbarem Luftzutritte versehenen, an den drei Füſsen des Apparates befestigten
weiten Messingring m, in welchen in Abständen von 3cm Löcher von 2 bis 3mm Durchmesser gebohrt sind. Der etwa 5l
haltende Raum läſst sich auf diese Weise sehr leicht auf 300° erhitzen, selbst wenn
er unten offen bleibt. Um dies zu erreichen, ist es aber wesentlich, daſs die
Zwischenräume zwischen den einzelnen Cylindern, durch welche die Flammengase gehen,
nicht weiter als etwa 10min sind und daſs der
äuſserste Cylinder f noch einen Schutzmantel aus einem
schlechten Wärmeleiter, z.B. Asbestpappe, erhält.
Will man aus diesem Apparate Destillationen ausführen, so wird der Kolben von unten
mit dem Halse durch den Ansatz k geschoben.
Zur Bestimmung des specifischen Gewichtes von
Flüssigkeiten saugt W. Nicol (Chemical News, 1883 Bd.
47 S. 85) den durch Fig. 17
Taf. 13 veranschaulichten Apparat A bis zur Marke c voll und wiegt in bekannter Weise. Um das specifische
Gewicht von festen Stoffen damit bestimmen zu können, erhält A einen seitlichen Ansatz.
Tafeln