| Titel: | Neuere Apparate für chemische Laboratorien. |
| Fundstelle: | Band 254, Jahrgang 1884, S. 67 |
| Download: | XML |
Neuere Apparate für chemische
Laboratorien.
Patentklasse 42. Mit Abbildungen im Texte und auf
Tafel 7.
Neuere Apparate für chemische Laboratorien.
Der Probestecher für chemische Producte von H.
Angerstein in Schalke (* D. R. P. Nr. 26680 vom 29. August 1883) besteht aus einer
unten scharf zugespitzten, runden, oben mit einem stärkeren Kopfe versehenen
Eisenspindel A (Textfigur
1). Das schmiedeiserne Rohr B ist unten auf ⅖
seiner Länge seitlich aufgeschlitzt. Beim Gebrauche wird die Spindel A in das Rohr B
eingeführt, das Ganze durch Hammerschläge auf den Spindelkopf in das zu bemusternde
Faſs bis zum Bunde b eingetrieben, nunmehr die Spindel
A herausgezogen, der Stift C in das Auge c des Rohres B eingeführt und damit letzteres in einer Richtung
mehrmals um seine Achse gedreht. Hierbei füllt sich das Rohr B an der seitlich aufgeschlitzten Stelle mit der Probe aus dem Fasse. Beim
Herausziehen des Rohres B gelingt es, diese Probe aus
der Masse, welche alle von der aufgeschlitzten Stelle durchbohrten Schichten
enthalten, auſserhalb des Fasses zu sammeln, während bei den bisherigen
Probestechern im Wesentlichen nur Proben vom äuſseren Rande des Fasses erhalten
wurden.
Fig. 1., Bd. 254, S. 68
Fig. 2., Bd. 254, S. 68
Currier empfiehlt im American
Druggist, 1884 Bd. 116 S. 25 zur Herstellung eines Scheidetrichters in das an einem Ende zugeschmolzene Glasrohr g (Textfigur 2) mittels
einer mit Terpentinöl und Campher befeuchteten Feile eine Oeffnung zu erzeugen und
dieses Rohr dann in den durchbohrten Kork e im Trichter
t zu schieben, so daſs zunächst diese seitliche
Oeffnung durch den Kork geschlossen wird. Um nun die untere Flüssigkeitsschicht
abzulassen, schiebt man das Rohr g in die Höhe, so daſs
die Oeffnung über dem Korke frei wird.
Fig. 3., Bd. 254, S. 68
V. Meyer (Berichte der deutschen
chemischen Gesellschaft, 1883 S. 3000) hängt den in Textfigur 3 skizzirten Trichter über die Abdampfschalen. In Folge
dessen entweicht die verdampfende Säure zum gröſsten Theile, während der durch
Condensation niedergeschlagene Theil in dem umgebogenen Rande des Trichters sich
sammelt und von Zeit zu Zeit weggenommen wird.
V. Meyer beschreibt ferner a. a. O. 1884 S. 478 einen
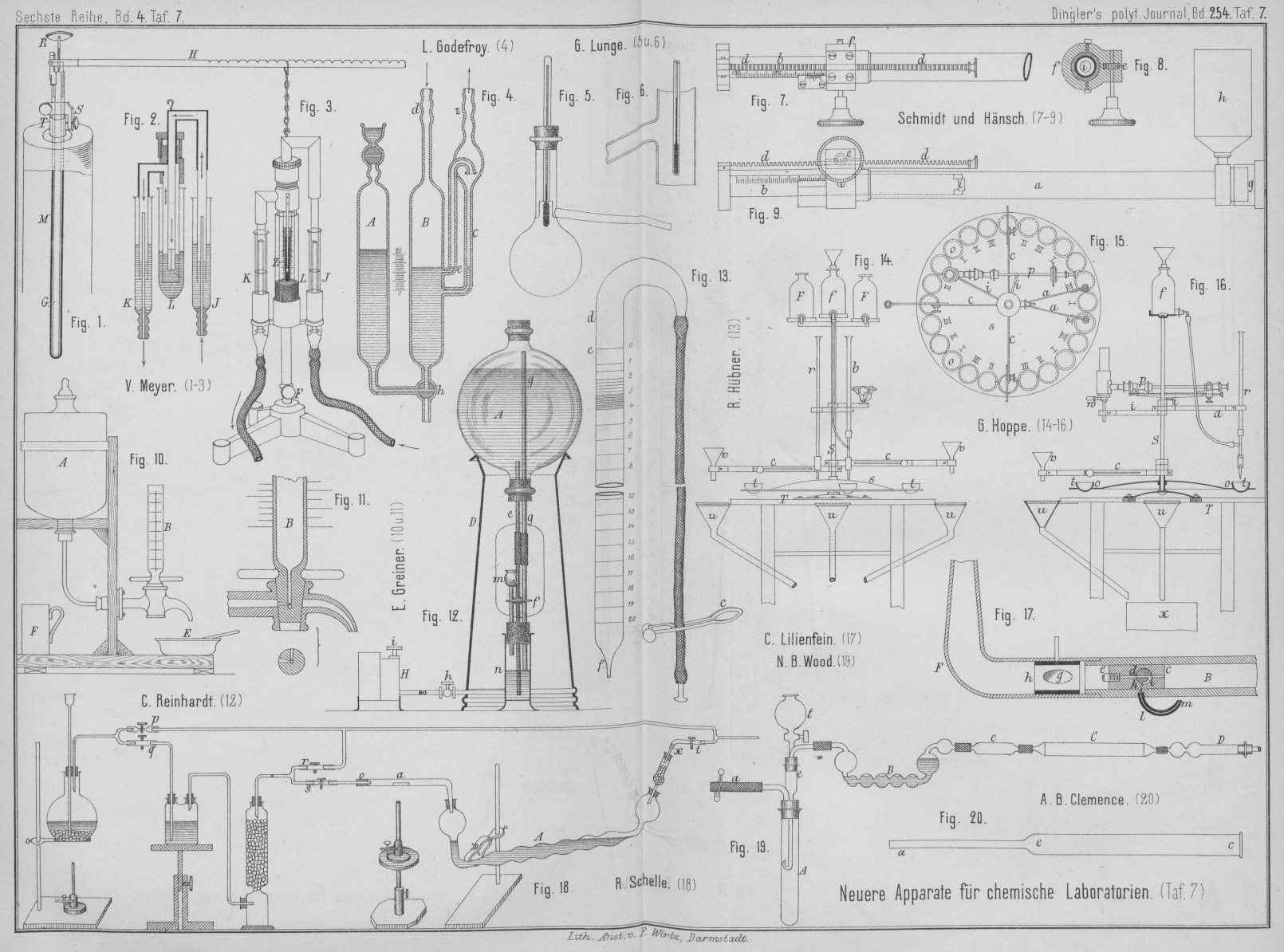
empfindlichen Temperaturregulator. In der 30cm langen Messingröhre M (Fig.
1 Taf. 7), welche durch eine verstellbare Verschraubung S an das Rohr T befestigt
ist, steckt der Glasstab G. Um den Unterschied in der
Ausdehnung des Glases gegen Messing zu vergröſsern, ist der Glasstab G mit einer Kappe versehen, aus welcher beiderseits ein
kleiner Stift wagerecht hervorragt. Auf diesem Stifte ruht 5mm von seiner Drehungsachse der an diesem Ende
gabelförmige, 40cm lange Hebel H, welcher mit einer Centimetertheilung und oben mit
entsprechenden Einschnitten versehen ist. In letztere greift der Haken einer Kette
ein, an welcher ein aus Glas und Messing hergestellter, dem oberen Theile des Kemp-Bunsen'schen Regulators nachgebildeter Apparat
(Fig. 2
und 3 Taf. 7)
hängt. Das Leuchtgas tritt in der Richtung der Pfeile durch zwei mit Glycerin
gesperrte Glasröhren J und K aus der Leitung ein und zum Brenner aus, so daſs der an der Kette
hängende Theil des Apparates ohne Reibung frei beweglich ist und seine
Verschiebungen mittels des Zeigers Z an einer auf das mittlere Glasrohr
L eingeätzten kleinen Millimeterskale abgelesen
werden können. Dieses Rohr L ist mit J und K zusammen in eine
auf einem Träger F verstellbare Fassung eingesetzt. L enthält Quecksilber, in welches das untere, aus einem
dünnen, bis auf einen feinen Schlitz zusammengebogenen Eisenbleche gebildete Ende
des Gaszuleitungsrohres eintaucht, welches je nach seiner Stellung durch den
seitlichen Schlitz mehr oder weniger Gas austreten läſst. Das Gas darf nicht
zwischen Glas und Eisen, sondern nur durch den 0,1 bis 0mm,2 weiten Schlitz austreten.
Stellt man nun den Träger F und mittels der Schraube R den Hebel H so, daſs der
Schlitz zum gröſsten Theile aus dem Quecksilber hervorragt, und entzündet das durch
den beschriebenen Regulirapparat zum Luftbade strömende Leuchtgas am Brenner
desselben, so wird in dem Maſse, als sich die Röhre M
erwärmt, der Glasstab G verhältniſsmäſsig verkürzt. Der
auf seiner Kappe ruhende Hebel sinkt und mit demselben auch der angehängte
Regulirapparat, so daſs der eiserne Schlitz tiefer in das Quecksilber eintaucht und
weniger Gas durchströmen läſst. Durch Drehen der die Kappe des Glasstabes
durchsetzenden, mit ihrem Ende auf dem Stabe ruhenden feinen Schraube R hebt man die Kappe und damit auch den Hebel H wieder, bis die Temperatur des Luftbades der
gewünschten oberen Grenze bis auf etwa 10 bis 20° nahe gekommen ist. Alsdann wird
durch Rückdrehen der Schraube R der Gaszufluſs auf das
Aeuſserste eingeschränkt. Damit derselbe nicht ganz aufhören könne, gieſst man
zweckmäſsig in die Röhre L nur so viel Quecksilber,
daſs der Spalt noch nicht ganz eintaucht, wenn die Messingfassung des
Zuleitungsrohres schon auf den oberen Rand des Glases L
aufstöſst, so daſs das Rohr nicht weiter einsinken kann. Durch feines Einstellen der
Schraube R läſst sich schlieſslich die gewünschte
Temperatur genau erreichen. Bei einer Länge von je 300mm beträgt also bei einer Erwärmung um 100° der Unterschied der
Längenausdehnung der Messingröhre M und des Glasstabes
G 0mm,3 oder für
1° nur 0mm,003. Da nun der Glasstab an einem
Hebelarme von nur 5mm wirkt, der Regulirapparat
aber an einem bis zu 400mm langen Hebel hängt, so
kann diese kleine Bewegung 80fach vergröſsert werden, so daſs für jede
Temperatursteigerung um nur 1° der eiserne Spalt um 0mm,24 aus dem Quecksilber herausgehoben wird. Ist der Spalt 30mm lang und 0,1 bis 0mm,2 breit, so entspricht seiner ganzen Länge bei dem Luftbade ein
Temperaturunterschied von reichlich 300°; d.h. wenn der Spalt fast ganz über dem
Quecksilber steht, so steigt die Temperatur des Bades über 300°. Durchschnittlich
gibt also eine Hebung von 0mm,1 schon eine
Steigerung von 1°.
Braucht man längere Zeit hindurch ein und dieselbe Temperatur, so kann man
vortheilhaft die bekannten Giroud'schen Rheometer (vgl.
1866 181 * 349. 1874 212 *
458) verwenden.
L. Godefroy (Annales de Chimie
et de Physique, 1884 Bd. 1 S. 140) füllt das Rohr A seines in Fig. 4 Taf. 7
dargestellten Druckregulators für fractionirte
Destillationen im luftverdünnten Räume mit trockenem Quecksilber. Die
Ansätze z und d werden mit
der Luftpumpe bezieh. mit dem Destillationsapparate verbunden. Das Quecksilber tritt
dann durch den Dreiwegehahn h nach B über und steigt bis zur Oeffnung e. Die von d kommende Luft
sucht dagegen das Quecksilber wieder herunterzudrücken, so daſs die Oeffnung von e in Zwischenräumen frei wird und Luft zur Pumpe
entweichen kann. Das dabei mit übergerissene Quecksilber flieſst durch Rohr c zurück. Man erhält so einen dem Höhenunterschiede des
Quecksilberstandes in A und B entsprechenden Druck.
G. Lunge (Chemische
Industrie, 1884 S. 150) untersuchte die verschiedenen Verfahren der fractionirten Destillation zur Werthbestimmung
von chemischen Producten. Verfasser erinnert daran, daſs ein und dieselbe
Flüssigkeit unter verschiedenen Bedingungen durchaus abweichende Siedepunkte zeigt.
Hierauf ist nicht nur der Barometerstand und die absolute Genauigkeit des
Thermometers von Einfluſs, sondern auch das Material des Destillationsgefäſses,
dessen Gestalt, diejenige des Thermometers, vor Allem die Stelle, welche das
letztere im Verhältnisse zum Abzugsrohre für die Dämpfe einnimmt, die Schnelligkeit
der Destillation u. dgl. Kein Wunder, daſs häufig Käufer und Verkäufer ganz
abweichende Ergebnisse erhalten. Hier kann nicht einmal ein dritter, unparteiischer,
durch seinen wissenschaftlichen Ruf anerkannter Chemiker endgültig entscheiden; denn
es handelt sich eben nicht um wissenschaftliche Methoden, welche prinzipiell bei
richtiger Ausführung nur eine Endzahl ergeben können,
wie etwa beim Titriren einer Soda, sondern um erfahrungsmäſsige Proben, welche nur
übereingekommenermaſsen feststehen und bei denen die geringste Abweichung an den
Versuchsbedingungen sofort das Ergebniſs verändert.
Bei den Arbeiten der Commission, welche der Verein für
chemische Industrie zur Vereinbarung von Methoden
für Handelsanalysen niedergesetzt hat, stellte es sich bald heraus, daſs
die Untersuchung von Handelsproducten durch fractionirte Destillation besonders
wichtig ist. Nach den von Lunge bei den
Vereinsmitgliedern eingezogenen Erkundigungen werden von den Verbindungen der feiten Reihe nur Aethylalkohol, Methylalkohol, Aldehyd,
Eisessig und Erdölproducte durch Destillation untersucht. Weitaus wichtiger ist die
fractionirte Destillation für aromatische Verbindungen:
Benzole aller Art (einschlieſslich Toluol und Xylol), Nitrobenzol u. dgl.; Anilin,
Toluidin, Xylidin, Cumidin, sogen. Echappées, Dimethylanilin, Diäthylanilin,
Benzylchlorid, Benzotrichlorid, Benzaldehyd, Chinolin, Chinaldin, Carbolsäure,
Naphtalin.
Die befragten Firmen destilliren ausschlieſslich in Glasgefäßen, mit Ausnahme von zweien, welche kupferne
Flaschen mit Glasaufsätzen benutzen. Man muſs hieraus wohl schlieſsen, daſs die
meisten Fabriken die Gefahr des Springens von Glasgefäſsen geringer achten als die
Bequemlichkeit der fortwährenden Beobachtung und die gröſsere Reinlichkeit, welche sich mit
denselben erreichen läſst. Ein Nachtheil der Glasgefäſse ist bekanntlich der, daſs
bei manchen Stoffen, vor Allem den Alkoholen, ein „Siedeverzug“ eintritt,
welcher die Destillationsergebnisse zu ganz regelwidrigen machen kann. Bei Benzol
und den meisten anderen hier zu behandelnden Stoffen bemerkt man dies nicht; auch
bei den Alkoholen kann der Siedeverzug durch Anwendung von Platinspiralen u. dgl.,
wie es einige Fabriken in bestimmten Fällen thun, aufgehoben werden.
Was die Form der Gefäſse betrifft, so
wendet keine Theerdestillation unter denen, welche geantwortet haben, die in England
gebräuchlichen Retorten mit in die Flüssigkeit eingesenkten Thermometern an. Von den
Farbenfabriken benutzen nur zwei dieses Verfahren und zwar ausschlieſslich auf das
aus England bezogene Rohbenzol. Es ist zu hoffen, daſs ein von den deutschen
Fabrikanten festgestelltes besseres Verfahren auch von den Engländern angenommen
würde.
Die deutschen Fabrikanten verwenden, mit den wenigen erwähnten
Ausnahmen, sämmtlich den sogen. Fractionirkolben. Mit
Festhaltung des Prinzipes, daſs das Quecksilbergefäſs des Thermometers sich im
Dampfe der eben überdestillirenden Flüssigkeit befinden soll, lassen sich doch zwei
Hauptklassen von Apparaten unterscheiden, nämlich solche, bei denen eine Verdichtung
des minder flüchtigen Theiles (sogen. Dephlegmirung) der Dämpfe erfolgt, und solche,
bei denen vielmehr die einmal entwickelten Dämpfe möglichst schnell abgeführt
werden. Die gewöhnlichen Fractionirkolben, wie dieselben in jedem chemischen
Laboratorium gebräuchlich sind, üben schon eine gewisse verdichtende Wirkung aus,
wenn man nicht eine bis zum Dampfrohre hinauf gehende Schutzhülle anwendet. Diese
Scheidung ist aber stets eine sehr unvollkommene und je nach der Länge des
Kolbenhalses und der äuſseren Temperatur schwankende. Wenig weiter in dieser sogen.
Dephlegmation gehen die Fabriken, welche im Kolbenhalse Erweiterungen anbringen.
Sehr wenige Fabriken verwenden einen für vollständige Dephlegmirung bestimmten
Aufsatz. In dieser Beziehung hat Kreis gezeigt, daſs
der theuere, durch seine Höhe lästige und äuſserst zerbrechliche Le Bel-Henninger'sche Aufsatz vollkommen durch das
einfache und billige Glasperlenrohr von Hempel ersetzt
werden kann und daſs man selbst mit kleinen Mengen von 50cc recht gute Erfolge erreicht. Mit gewöhnlichen
Fractionirkolben führt nach diesen Untersuchungen nur eine gröſsere Reihe von
Fractionirungen (12) zum Ziele einer annähernd vollständigen Trennung, z.B. von
Benzol und Toluol; auch bei Würtz'schen Zweikugelkolben
braucht man noch 6 Destillationen; man kommt dagegen mit einer Hempel'schen Röhre schon durch eine einzige
Destillation zu einem fast ebenso guten Ziele. Aehnlich wirkt auch der etwas
umständliche Linnemann'sche Aufsatz.
Zwei Fabriken erstreben dagegen eine vollständige Abführung einmal gebildeter Dämpfe. Das Dampfabzugsrohr ist
nämlich, wie Fig.
5 Taf. 7 zeigt, dicht über dem Bauche des Kolbens angesetzt, wo auch ein
inneres Rohr so angeschmolzen ist, daſs die im Halse des Kolbens sich verdichtende
Flüssigkeit nicht in den Bauch desselben zurücktropfen kann, sondern durch das
Dampfabzugsrohr in den Kühler fortläuft. Das schmale, cylindrische Quecksilbergefäſs
des Thermometers befindet sich in der Mitte des Stutzens und wird durch diesen vor
Abkühlung von auſsen geschützt. Der für die Beurtheilung maſsgebende Grad des
Thermometers befindet sich gerade über dem Korke, so daſs die Quecksilbersäule so
gut wie ganz im Dampfe ist. Dies erfordert natürlich eigene Thermometer mit
verschieden hoch anfangender Theilung für jede einzelne Flüssigkeit.
Handelt es sich um Stoffe, welche nur einen einzigen werthvollen
Bestandtheil von bestimmtem Siedepunkte enthalten, während alles darunter oder
darüber Siedende als Verunreinigung angesehen werden muſs, z.B. Methylalkohol,
Aldehyd, Dimethylanilin, Benzylchlorid, Benzaldehyd u. dgl., ferner auch reines Benzol, Toluol, Xylol, Nitrobenzol, Anilin, so
scheint diese letztere Vorrichtung empfehlenswerth, wegen der Verhütung alles
Zurücktropfens einmal destillirter Stoffe; namentlich bei hochsiedenden Stoffen hat
dies den Vorzug, einer Zersetzung durch zu lang dauernde Erhitzung vorzubeugen.
Anders verhält es sich bei Gemengen, von verschiedenen Stoffen,
deren Siedepunkte nicht weit von einander liegen und bei denen nicht nur ein
einzelner Gemengtheil, sondern mehrere derselben von Werth sind, so daſs es darauf
ankommt, die Menge der einzelnen Bestandtheile zu ermitteln, weil dieselben einen
ungleichen Werth besitzen, z.B. Erdölbenzine, Naphta, Leuchterdöl, die verschiedenen
Handelsbenzole, Nitrobenzole, Anilinöle, sogen. Echappées u. dgl. Bei Erdölproducten
wird man mit einer gewissen Willkür bestimmte Anfangs- und Endpunkte für das
Thermometer festsetzen müssen, wobei freilich alle Einzelheiten des anzuwendenden
Destillationsapparates ebenso genau wie sonst festzustellen sind. Auch wird man
hierbei das specifische Gewicht ebenso wie den Siedepunkt als Werthmesser mit
heranziehen müssen.
Beim Handelsbenzol wird nicht einmal der Siedepunkt des reinen
Benzols, Toluols u. dgl. zu Grunde gelegt, sondern man fragt danach, wie viel
Procent bei 100° übergehen, während bei Nitrobenzol, Anilin, Carbolsäure u.a. doch
der eigentliche Siedepunkt der reinen Verbindungen zu Grunde gelegt wird. Der
Handelsgebrauch beim Benzol schreibt sich augenscheinlich davon her, daſs bei dem
bekannten rohen englischen Prüfungsverfahren das Quecksilber im Thermometer
erheblich über den Siedepunkt der gerade destillirenden Flüssigkeit steigen muſs; dieser Gebrauch ist aber einmal so fest eingeführt,
daſs es schwer sein würde, denselben zu ändern. Auch scheint dies nicht unumgänglich
nothwendig, da ja hier, sowie bei den Anilinölen u. dgl., auch die höher siedenden
Verbindungen ihren Werth besitzen und man nur eben einen Gradmesser für diesen
erlangen will. Aber es ist doch kaum zu bezweifeln, daſs wenigstens dem Käufer weit
mehr gedient wäre, wenn er wirklich genau oder nahezu genau wüſste, wie viel
wirkliches Benzol, Toluol, Anilin, Toluidin u. dgl. in den von ihm zu verwendenden
Producten enthalten ist. Wollte man diese gemischten Producte so destilliren, daſs
die einzelnen Bestandtheile möglichst vollständig getrennt werden, so könnte man
z.B. die Glasperlenröhre anwenden. Bei niedrig siedenden Flüssigkeiten muſs man dann
aber stärkere Kühlung, also mehr Kugeln, ein längeres Glasperlenrohr u.s.w. als bei
höher siedenden anwenden; bei den letzteren muſs man sogar das Rohr oft noch mit
einer Schutzhülle gegen zu schnelle Abkühlung versehen, so daſs man also nicht
denselben Apparat für alle möglichen Stoffe verwenden kann.
Die gründlichste Lösung der Schwierigkeit würde vielleicht darin
bestehen, daſs man bei der Destillation im Kleinen die Bedingungen einer
fabrikmäſsigen Rectification genauer nachahmt, als dies durch bloſse Luftkühlung
geschehen kann. Lunge hat sich schon seit längerer Zeit
in seinem Laboratorium für die Uebungen der Praktikanten in Darstellung von
Präparaten eines Apparates bedient, welcher sich wohl zu obigem analytischen Zwecke
ausbilden lieſse. Die Dämpfe entweichen bei demselben durch ein längeres, schief
ansteigendes Glasrohr, welches in einem Blechkasten liegt. Der Kasten ist mit
Wasser, für höher siedende Flüssigkeiten mit Chlorcalciumlösung, Paraffin u. dgl.
gefüllt und wird auf bestimmte Temperatur erwärmt. Von da geht das Glasrohr
möglichst scharf in einen gewöhnlichen absteigenden Kühler über. Das Thermometer im
Destillationskolben kann hier allenfalls entbehrt werden; jedenfalls aber muſs ein
solches in der Flüssigkeit des Blechkastens angebracht sein. Die Erhitzung des
Kastens sollte durch einen Thermoregulator geschehen, so daſs man eine bestimmte
Temperatur längere Zeit einhalten kann. Man würde also z.B. für Prüfung eines
Benzols auf 800 erwärmen; das Benzol selbst wird jetzt noch überdestilliren, das
Toluol wird zurückflieſsen. Wenn nichts mehr übergeht, wechselt man die Vorlage oder
liest nach Bedürfniſs einfach das Volumen des Destillates ab und erhitzt nun den
Blechkasten auf 110°, bis alles Toluol übergegangen ist u.s.f.
Bei diesem Apparate lieſse sich auch mit aller Leichtigkeit und
mit viel gröſserer Sicherheit als sonst die Benzolprüfling mit dem Fixpunkte 100°
ausführen, sogar unabhängig von Temperaturschwankungen. Man würde zu diesem Zwecke
den Kasten bedecken, auf den Deckel ein Kühlrohr setzen und das Wasser zum Kochen
bringen; das verdampfende Wasser flieſst immer wieder zurück. Da der Fixpunkt 100°
doch den Siedepunkt des Wassers bedeuten soll, so wäre diese Bedingung hier bei
beliebigem Luftdruck erfüllt, ohne daſs man den Barometerstand zu beobachten brauchte.
Selbstredend müſste durch genaue Versuche festgestellt werden, wie weit die Angaben
dieses Apparates mit denen der gewöhnlichen Apparate und der wirklichen
Zusammensetzung der Substanzen übereinstimmen; vermuthlich könnte man dieselben
keineswegs mit denen des englischen Retortenapparates unmittelbar vergleichen.
Die bestimmteste Vereinbarung über die Form des Destillirapparates
nutzt aber nichts, wenn nicht das zu verwendende Thermometer genau istVgl. F. Fischer: Chemische Technologie der
Brennstoffe, S. 19. und wenn dasselbe nicht genau an der
richtigen Stelle steht. Auſser der Prüfung des Punktes 100° mit Wasser und 218° mit
Naphtalin ist es sehr empfehlenswert, mit demselben Thermometer und sonst dem
gleichen Apparate eine ganz reine Substanz von möglichst nahem Siedepunkte wie die
zu untersuchende zu destilliren und sich nach den so erhaltenen Angaben zu richten.
Für Anilinproben z.B. destillirt man also zunächst chemisch reines Anilin, setzt die
dabei am Thermometer beobachtete Temperatur, was auch dabei herauskommen möge, als
normal und wiederholt gleich darauf die Arbeit mit dem zu prüfenden Anilinöl. Dabei
macht man sich natürlich auch von den Schwankungen des Barometerstandes unabhängig.
Bequem sind hierfür die Thermometer mit durch eine Schraube verstellbarer SkaleZu beziehen von F. Müller in Berlin,
Kanonierstraſse 1., welche mit Hilfe von Normalsubstanzen auf
bestimmte Hauptpunkte eingestellt werden. Man benutzt zwei Thermometer. Die Skale
des einen reicht von 40 bis 170; es wird mit Wasser eingestellt und dient z.B. für
Benzol, Toluol und Xylol. Das andere Thermometer geht von 100 bis 260° oder 140 bis
280°, wird für höher siedende Flüssigkeiten benutzt und seine Skale mit chemisch
reinem Anilin so eingestellt, daſs die Kuppe des Quecksilberfadens bei 182° steht,
wenn von 100cc des Anilins 60cc übergegangen sind.
Bei der Stellung des Quecksilbergefäſses kommt es weniger auf
völlige Gleichmäſsigkeit unter den verschiedenen Fabriken an, wenn man die
Thermometerskale in angegebener Weise vorher auf einen bestimmten Grad feststellt
und dann später das Thermometer genau an derselben Stelle anbringt. Unter den
verschiedenen jetzt gebräuchlichen Stellungen scheint diejenige das Mittel zu
bilden, bei welcher das obere Ende des Quecksilbergefäſses mit der Unterkante des
Dampfabzugsrohres zusammenfällt, wie Fig. 6 Taf. 7 zeigt. Das
Gefäſs selbst sollte nicht über 10mm lang
sein.
Die Mehrzahl der angefragten Fabriken bringt überhaupt keine Berichtigung für den Barometerstand an, wodurch ganz
erhebliche Abweichungen entstehen können. Einige beobachten wenigstens das Barometer
und hüten sich bei starker Abweichung vom Normalstande vor bestimmten Schlüssen;
oder sie helfen sich dann durch Destillation einer Normalprobe von bekanntem
Siedepunkte, Dieser Ausweg wird von mehreren der Fabriken überhaupt unter allen
Umständen ergriffen und von einer sogar die Skale des Thermometers jedesmal neu
eingestellt. Einige wenige Fabriken wenden Reductionstabellen an, die aber zum
Theile gewiſs unrichtig sind, da sie einfach die Zahlen für die Veränderung des
Siedepunktes des Wassers unter bestimmten Drucken auf die anderen Flüssigkeiten
übertragen, deren Dampfspannung sicher ganz andere Beziehungen zum Luftdrucke als
die des Wassers hat. Eine Fabrik besitzt Erfahrungstabellen, übt daneben aber noch
Destillation eines Typs. Da man eigentlich für jede einzelne in Rede kommende
Substanz besondere Siedepunktstabellen für die Schwankungen des Luftdruckes mit
physikalischer Genauigkeit ermitteln müſste und dann immerhin nur eines der störenden Elemente beseitigt hätte, so wird
sich aus diesem Grunde statt solcher Tabellen die jedesmalige Beobachtung des
Siedepunktes einer Normalflüssigkeit bezieh. Einstellung des Thermometers auf einen
Normalpunkt empfehlen, wie diese oben beschrieben ist. Das Barometer würde dann gar
nicht mehr abzulesen sein.
Zu berücksichtigen ist ferner, daſs der Ansatz des Abzugsrohres an dem Kolbenhalse nicht verengt sein sollte. Die
Länge und Weite des Kühlers, sowie dessen Neigungswinkel sind auch nicht
gleichgültig und sollten ebenfalls vereinbart werden, desgleichen die Schnelligkeit der Destillation. Gasbrenner und womöglich auch das
Destillationsgefäſs sind durch Asbestpappe oder Blech vor Zugluft zu schützen.
Wenn ein Fixpunkt als entscheidend angesehen wird, z.B. 100° für
Handelsbenzol, so gilt die (auch in der englischen Vorschrift vorkommende) Regel,
daſs man die Flamme ausdreht, sowie das Quecksilber über dem Striche erscheint, das
noch im Kühler Befindliche sich verdichten und nachtropfen läſst und erst dann
abliest. Will man nun noch die Procentigkeiten bei höheren Siedepunkten beobachten,
was ja gerade für Benzol sehr wichtig ist, so wird man die Flamme wieder anzünden
und noch bei beliebigen höheren Graden in ähnlicher eben beschriebener Weise nach
dem Abtropfen ablesen. Die meisten Fabriken wenden als Vorlagen Cylinder mit
Theilung an, in denen direkt abgelesen wird. Eine solche Vorlage hat ein Gestell mit
12 kreisförmig angeordneten Glasröhren von 15mm
Durchmesser in 0cc,1 getheilt, welche zum
Auffangen der Fractionen hinter einander unter das Ende des Kühlrohres geschoben
werden.
Für Benzole kommt auch das
specifische Gewicht der Fraction in Betracht. Bei 90 und bei 50procentigem Benzol
soll nichts unter 80° übergehen, bei 90procentigem Benzol nichts über 120°, bei 50 procentigem nichts über 130°. Beim
Schütteln mit 66° Schwefelsäure soll sich das Benzol gar nicht, die Schwefelsäure
nicht gleich färben und letztere soll nach 10 Minuten erst gelblich sein; später
wird dieselbe immer schwarz. Noch wichtiger ist das Verhalten gegen Salpetersäure
von 40° B. Beim Eingieſsen derselben in das Benzol sollen keine weiſsen Dämpfe
entstehen; beim Schütteln damit soll sich das Benzol nicht färben. Bei längerem
Stehen wird die Salpetersäure schwach röthlich; später wird die Säure farblos, die
rothe Farbe geht aber auf das Benzol über. Naphtalin
wird aus einer Retorte destillirt, wobei das Quecksilbergefäſs gegenüber dem
untersten Theile des Halses angebracht ist. Das zwischen 210 und 225° Uebergehende
wird aufgefangen. Benzaldehyd wird zweckmäſsig im
Kohlensäurestrome destillirt. Von Methylalkohol sollen
beim Sieden mit eingelegter Platinspirale 98,5 bis 99 Proc. von 66 bis 66,5°
übergehen. – Bei Erdöl für Leuchtzwecke werden nach Schenkel 200cc mit
Perlenröhre und Dreiwegerohr destillirt und zwar so, daſs in 1 Minute etwa 2cc übergehen. Bei 150° entfernt man die Flamme,
liest ab, entfernt nach 10 Minuten das Perlenrohr und steckt das Dreiwegerohr direkt
auf den Kolben, worauf man bis 300° weiter destillirt. Ein Erdöl, welches unter 150°
mehr als 5 Proc. Destillat abgibt, ist unzulässig, ein solches, das bis 300° nicht
90 Proc. liefert, ist von geringer Güte. Was von 150 bis 300° übergeht, ist als
„Normal-Erdöl“ (vgl. auch F. Beilstein 1883
250 170) anzusehen.
F. Schmidt
und Hänsch in Berlin (* D. R. P. Nr. 25439 vom 21. Juli 1883) verwenden für ihre
Polarisationsinstrumente eine Controlvorrichtung
für die Quarzkeile, welche auf dem Biot'schen Gesetze
beruht, wonach die Drehung der Polarisationsebene der Dicke der Schicht und der
Menge der gelösten Substanz proportional ist. Wie aus Fig. 7 bis 9 Taf. 7 zu entnehmen, ist
nur die eine Glasplatte am rechten Ende der Röhre a
fest, die andere Platte i sitzt aber an dem Ende einer
Röhre b, welche in a
wasserdicht hin- und hergeschoben werden kann. Zu diesem Zwecke geht von dem freien
Ende der Röhre b eine Zahnstange d aus, welche durch ein Getriebe e in der Fassung f der
Röhre a bewegt wird. Bei der Handhabung nimmt man
zunächst die Kappe g mit der Verschluſsplatte aus Glas
ab und zieht die Röhre b so weit wie möglich aus der
Röhre a; dann wird letztere mit der zu untersuchenden
Flüssigkeit gefüllt und hierauf durch Glasplatte und Kappe g wieder geschlossen. Man steckt nun das Gefäſs h mit Rohransatz in ein vorher durch einen Stöpsel verschlossenes Loch der
Röhre a, so daſs beim Verschieben der Röhre b
die aus a verdrängte Flüssigkeit in das Gefäſs h treten kann. Aus der an der Skale abgelesenen Länge
der Flüssigkeitssäule läſst sich nun durch Rechnung für eine bestimmte Flüssigkeit
leicht ermitteln, um wie viel die Flüssigkeitssäule verlängert oder verkürzt werden
muſs, um nach Verschiebung des Quarzkeilpaares wieder Farbengleichheit und gleiche
Beleuchtungsstärke zu erzielen.
G.
Hoppe in Bohrendorf (* D. R. P. Nr. 27287 vom 13. Oktober 1883) hat bei seinem Titrirapparate für Rübensäfte u. dgl. auf dem Tische
T (Fig. 14 bis 16 Taf. 7) ein
Stativ S mit drehbarer Scheibe s befestigt, in deren Oeffnungen o die
Titrirschalen t eingehängt werden. Um Verwechselungen
zu vermeiden, sind die Oeffnungen o nach einander mit
den drei römischen Ziffern I bis III entsprechend der ersten, zweiten und dritten
Saturation des Saftinhaltes der Schalen bezeichnet. Die Stativstange S trägt oben eine Flasche f zur Aufnahme der Probesäure, ein Gabelarm a
an einem Ende eine Bürette b mit Probesäure, am anderen
eine Röhre r mit Corallin. Drei in der Länge
verstellbare Arme c tragen je einen Trichter v zum Filtriren der drei verschiedenen Säfte. Unter
jedem dieser Trichter sind am Tische T Auffangtrichter
u angebracht, welche den überlaufenden Saft in ein
gemeinsames, unter dem Tische aufgestelltes Gefäſs x
überleiten.
Der zu untersuchende Saft wird in einem der Trichter v
filtrirt, von welchen drei, je einer für einen der Säfte, vorhanden sind. Den
durchfiltrirten Saft läſst man in ein Gefäſs von 10cc Inhalt eintropfen, gieſst nach erfolgter Füllung den Saftinhalt in die
dazu bestimmte Schale t und stellt die letztere
zunächst unter die mit Corallinflüssigkeit gefüllte Röhre r ein. Von dieser Flüssigkeit setzt man dem Safte so viel zu, bis derselbe
roth gefärbt erscheint. Hierauf stellt man die betreffende Schale unter die
Probesäure enthaltende Bürette b ein und läſst davon so
viel zutröpfeln, bis der Saft wieder diejenige Farbe annimmt, welche derselbe vor
dem Zusätze von Corallinflüssigkeit hatte. Aus der Menge der abgelaufenen Probesäure
ersieht man dann, ob die Säfte genügend saturirt sind.
Die ebenfalls von dem Stative S getragenen Flaschen F dienen zur Bestimmung des Zuckergehaltes im
Abdruckwasser und in den ausgelaugten Schnitzeln; ferner ist an dem Stative mittels
zweier Arme i ein Polarisator p mit der Spitze w für den Anschluſs eines
Gummischlauches zur Speisung des Gasbrenners befestigt.
Der Titrirapparat von E. Greiner in
Stützerbach, Thüringen (* D. R. P.
Nr. 26830 vom 11. August 1883) soll namentlich zur Bestimmung der Alkalität in den Saturationssäften
dienen. Die Skale der Bürette zeigt rechts die Eintheilung in Cubikcentimeter, links
die entsprechende Kalkmenge. Man füllt die Flasche A
(Fig. 10
und 11 Taf.
7) mit Normalsäure, miſst mit dem Glase F 100cc Saturationsflüssigkeit, gibt dieselbe in die
Schale E und färbt mit einigen Tropfen Lackmustinctur
blau. Dreht man nun das
Küken der Hahnbürette B, so flieſst die Normalsäure zu
und steigt bis zum Nullstriche. Dann wird durch Rechtsdrehen weiteres Zuflieſsen
verhindert. Dreht man noch weiter nach rechts, so kann die Säure durch den Hahn
abflieſsen und mischt sich tropfenweise mit dem gemessenen Safte in der Schale E. Tritt die violette Färbung ein, so dreht man das
Küken zurück. Die Skale gestattet nun die direkte Ablesung der Alkalität in
Procent,
R.
Hübner in Jena (* D. R. P. Nr. 27505 vom 14. November 1883) verwendet als Pipettbürette ein unten zu einer feinen Oeffnung
ausgezogenes, entsprechend getheiltes Glasrohr, welches, wie aus Fig. 13 Taf. 7 zu sehen,
oben gebogen ist. Dasselbe wird mit dem oberen, nicht getheilten Ende de in einen Halter geschraubt, dann das Gefäſs mit der
Normallösung so weit von unten über dasselbe geschoben, daſs die ausgezogene Spitze
f, je nach der gewünschten Menge Lösung, genügend
weit in die Flüssigkeit taucht. In dieser Stellung saugt man unter Lüftung des
Quetschhahnes c Luft durch den Schlauch und so die
Flüssigkeit in dem Rohre bis zu jedem beliebigen Punkte, auf welchem dieselbe
selbstverständlich stehen bleibt, sobald man den Quetschhahn wieder wirken läſst.
Das Austreten der Flüssigkeit aus der so gefüllten Bürette kann nun durch den
Quetschhahn c, also durch allmählichen Luftzutritt zu
dem luftverdünnten Räume, genau so geregelt werden wie bei jeder anderen
Bürette.
Th. Pusch empfiehlt im Archiv
der Pharmacie, 1884 Bd. 222 * S. 22 eine Cylinderbürette, bestehend aus einem etwa 20cm hohen, 1cm weiten getheilten
Glascylinder mit Fuſs und Ausguſsschnabel.
Fig. 4., Bd. 254, S. 76
A. G.
Martin in St. Denis (* D. R. P. Nr. 26920 vom 23. Oktober 1883) empfiehlt, zur sicheren Beobachtung des Standes von Flüssigkeiten
eine farbige Linie a auf weiſsem Grunde b anzubringen, wie Textfigur
4 andeutet, um durch Reflexwirkung die Flüssigkeitsoberfläche besser
erkennbar zu machen.
Fig. 5., Bd. 254, S. 76
G. Loges verwendet nach der Chemikerzeitung, 1884 S. 69 zur Bestimmung der
Härte des Wassers mit Seifenlösung ein etwa 4mm weites, 15cm langes Messingrohr,
dessen 1cm weite Kugel mit 30 feinen
Durchbohrungen versehen ist (vgl. Textfigur 5). Zur
Ausführung der Härtebestimmung befestigt man Blaserohr und Bürette für
Titerflüssigkeit dicht neben einander an einem Halter. Man bringt dann 40cc bezieh. eine geringere, mit destillirtem Wasser
auf 40cc aufgefüllte Menge des zu untersuchenden
Wassers in ein Becherglas, dessen Gröſse so zu wählen ist, daſs die
Flüssigkeitsschicht 2 bis 3cm hoch ist. Indem man
nun einen kräftigen, möglichst gleichmäſsigen Luftstrom durch das Rohr treibt,
bringt man durch Heben des Glases die Kugel in das Wasser und regelt mit der anderen
Hand den Zufluſs der Seifenlösung aus der Bürette. Durch die vielen heftig durch das
Wasser gepreſsten
Luftbläschen entsteht etwas Schaum, welcher sich aber nicht über die
Wasseroberfläche erhebt. Sobald ein Ueberschuſs der Seifenlösung zugesetzt ist,
steigt der Schaum plötzlich in groſsen Blasen bis über den Rand des Becherglases.
Durch Senken des Becherglases entfernt man die Kugel aus dem Wasser, der Schaum
sinkt sofort zusammen. Wird durch zweites Anblasen wieder der hochsteigende Schaum
erzeugt, so ist die Reaction als beendet anzusehen.
Das von G. Loges verwendete Zersetzungsgefäſs zum Scheibler'schen Kohlensäure-Apparate besteht aus
einem etwa 15cm hohen Pulverglase, dessen
durchbohrter Kautschukstöpsel ein mit seitlicher Oeffnung e (Textfigur 6) versehenes Glasgefäſs g trägt. Man bringt nun in gewöhnlicher Weise die zu
zersetzende Substanz in die Flasche, füllt das innere Glasgefäſs durch einen
Trichter mit gebogenem Rohre bis zur Marke mit Salzsäure und läſst nach
Zusammensetzung des Apparates die Säure durch seitliches Neigen des Gefäſses
ausflieſsen.
Fig. 6., Bd. 254, S. 77
N. B. Wood (Scientific American
Supplement, 1883 S. 6553) bringt zur Bestimmung des
Kohlenstoffes in Eisen und Stahl die Probe in das Rohr A (Fig. 19 Taf. 7) und läſst
aus dem Trichterrohre t langsam Säure zutropfen. Die
entwickelten Gase durchziehen das mit Asbest oder mit durch Schwefelsäure
befeuchtete Bimssteinstücken gefüllte Rohr e, gehen
durch das mit alkalischer Bleilösung gefüllte Kugelrohr B, welches den Schwefelwasserstoff zurückhält, dann durch das
Chlorcalciumrohr c in das mit Kaliumdichromat gefüllte
Verbrennungsrohr C, welches durch drei Bunsen'sche Brenner erhitzt wird, schlieſslich durch
ein Chlorcalciumrohr D, einen Liebig'schen Kaliapparat, nochmals durch ein Chlorcalciumrohr zu einem
Sauger. Hat die Gasentwickelung nachgelassen, so öffnet man den Quetschhahn auf a und zieht Luft hindurch. Aus der Gewichtszunahme des
Kaliapparates berechnet man den gebundenen Kohlenstoff, während der freie
Kohlenstoff durch Abfiltriren der in A erhaltenen
Lösung und der Schwefel in der Bleilösung des Rohres B
bestimmt werden kann.
A. B. Clemence (Engineer,
1883 Bd. 56 S. 387) löst zur Bestimmung des Kohlenstoffes in
Stahl 3g der Probe in 36g Ammoniumkupferchlorid und 120cc Wasser, filtrirt durch den in Fig. 20 Taf. 7
gezeichneten Platintrichter, welcher bei e einen
Asbestpfropfen enthält, wäscht aus und trocknet bei 150 bis 170°. Nun setzt man bei
c einen Stopfen mit Glasrohr zum
Sauerstoffgasometer ein, verbindet das andere Ende a
mit dem Absorptionsapparate und erhitzt die Stelle e
mit einer Gasflamme, so daſs der Kohlenstoff verbrennt und in bekannter Weise
bestimmt wird.
B. Schelle (Berg- und
Hüttenmännische Zeitung, 1883 S. 589) läſst zum Aufschlieſsen der Erze mittels Chlor das durch Schwefelsäure oder Chlorcalcium getrocknete
Chlorgas durch das mit Quetschhähnen r und s (Fig. 18 Taf. 7) in das
12mm weite Aufschlieſsungsrohr a treten, welches aus schwer schmelzbarem Glase
hergestellt ist. Das Absorptionsrohr A ist so in einem
Retortenhalter gefaſst, daſs der ganze Apparat von o
bis x schnell und leicht fortgeschafft und durch einen
anderen ersetzt werden kann. Auf A ist noch ein
kleines, mit feuchten Glasperlen oder Glaswolle gefülltes Absorptionsröhrchen x von bekannter Form und Gröſse aufgesetzt.
Nachdem das Absorptionsrohr A bei richtiger Stellung bis
zu den zwei letzten Kugeln mit Weinsäure haltiger verdünnter Salzsäure gefüllt ist,
wozu man zweckmäſsig die Hälfte Chlorwasser hinzufügt, und bei geöffnetem
Quetschhahne r und geschlossenem Hahne s die Luft aus dem Apparate verdrängt ist, wird das
abgewogene trockene Erzpulver in einem Porzellanschiffchen in das
Aufschlieſsungsrohr a hineingebracht, verschlossen und
mit dem Gabelrohre in Verbindung gesetzt. Nach Oeffnen der Hähne s und t und Schlieſsen von
r tritt das Chlorgas in den Apparat und die
Zersetzung beginnt. Nach beendigter Zersetzung, welche zuletzt durch Erhitzen des
Schiffchens beschleunigt wird, wird das Sublimat mit der Flamme bis über die
Krümmung hinausgetrieben, der Apparat nach geeigneter Stellung der Hähne r, s und t ausgeschaltet,
das Schiffchen mittels eines Platindrahtes herausgezogen und das Rohr verstopft. Der
ganze Apparat wird jetzt durch einen anderen ersetzt.
Es ist zuweilen schon während der Arbeit oder doch bei Beginn derselben nothwendig,
den Kolben frisch zu füllen, bei welcher Gelegenheit es dann längere Zeit dauert,
bis die Luft durch die schon mit Chlor gefüllten Trockenapparate getrieben und
dieselben wieder mit Chlor gefüllt sind. Bei obiger Anordnung hingegen wird einfach
die, Luft aus dem Kolben bei geöffnetem Quetschhahne p
und geschlossenem Hahne q verdrängt und hierauf nach
umgekehrter Stellung der Hähne mit den Trockenapparaten in Verbindung gebracht.
Die von C. Lilienfein in Stuttgart construirte Lampe für niedrig siedendes Erdöl besteht nach der Zeitschrift für analytische Chemie, 1884 S. 35 aus
einem Metallgefäſse und einem in dasselbe eingeschraubten, der Bunsen'schen Gaslampe nachgebildeten Brenner B (Fig. 17 Taf. 7), welcher
einen Docht enthält und von dem Metallgefäſse aus mit dem Brennstoffe gespeist wird.
Die Hahnvorrichtung besteht aus einem Metallstücke c
mit feiner Durchbohrung zur Ausströmung des Brennstoffes. Diese Bohrung sowie noch
ein zweiter senkrecht darauf stehender Kanal ist durch Hahn d absperrbar. In das Metallstück c ist ein
kleiner Einsatz e mit einer feinen Durchbohrung für den
Austritt des Brennstoffes eingeschraubt; letzterer strömt daraus in ein an B angeschraubtes Röhrenstück F, welches in einer Rundung nach aufwärts gebogen und mit Luftzuglöchern
g versehen ist, sowie mit einer zur beliebigen
Verkleinerung derselben geeigneten Hülse h, ganz
entsprechend wie beim Röhreneinsatze der Bunsen'schen Lampe. Statt
dieses Röhrenstückes F läſst sich auch ein anderes nach
Art der Gebläselampen eingerichtetes anschrauben. Setzt man dann die Vorrichtung mit
einem Blasebalge oder einem Gasometer in Verbindung, so erhält man eine starke
Gebläseflamme. Die Vergasung des Erdöles geschieht dadurch, daſs man vor dem
Anzünden der Lampe während einiger Minuten mit einer anderen Flamme das Rohr B anwärmt; dann besorgt
dieses eine Zweigleitung, welche aus einer Rinne i im
Eisenstifte d, einer Durchbohrung h im Stücke c und einem
damit verbundenen Röhrchen l besteht, dessen Mündung
m unter der Röhre B
gegen das Gefäſs hin endet. Bei geöffnetem Hahne sind auch die diese Zweigleitung
zusammensetzenden Kanäle verbunden, so daſs also das durch Vorerwärmen vergaste
Brennmaterial nicht nur durch den Brennereinsatz e
strömt, sondern auch durch die Zweigleitung und an deren Mündung m angezündet werden kann. Die dort brennende Flamme
besorgt dann die weitere Vergasung des Brennstoffes.
Um nun auch noch die durch den flüssigen Zustand des Brennstoffes bedingte, im
Vergleiche zu Leuchtgaslampen umständliche Arbeit eines jedesmaligen Vorerwärmens
nach kurzer Unterbrechung der Flamme zu beseitigen, ist die Zweigleitung für die
Vergasung derart mit der Hauptleitung durch den Hahn verbunden, daſs man letztere
ganz abschlieſsen, also die Hauptflamme abstellen kann, während die Zweigleitung
noch offen bleibt, so daſs das Flämmchen bei m
weiterhin fortbrennt und zwar beliebig verkleinert. Dadurch dauert die Erwärmung und
Vergasung des Brennstoffes fort, so daſs beim Wiederöffnen des Hahnes auch die
Hauptflamme sogleich wieder angezündet werden kann.
Ebenfalls für solche Laboratorien, welche noch kein Leuchtgas zur Verfügung haben,
dient die von C. Bernhardt (daselbst S. 40) angegebene
Spirituslampe. Der etwa 2l,5 fassende Spiritusbehälter A (Fig. 12 Taf. 7) ruht auf
dem Zinkblechständer D, welcher vorn einen gröſseren
Ausschnitt besitzt, um den Hahn f erreichen zu können.
Die Flamme der aus starkem Messingbleche hergestellten Spirituslampe H mit doppeltem Luftzuge ist durch Schraube i regulirbar. Bei geschlossenen Hähnen h und f wird die Kugelt
mit Spiritus gefüllt, ohne daſs solcher in das Rohr g
gelangt, worauf man die Kugel oben schlieſst. Der Stand des Spiritus im Behälter n und somit auch in der Lampe H ist durch die höhere oder tiefere Stellung des Luftrohres g bedingt. Hat man dieses passend eingestellt, so
werden die Hähne f und h
geöffnet, so daſs durch Rohr m nach n und von da durch g Luft
in den Spiritusbehälter A tritt; der Spiritus flieſst
in Folge dessen so lange durch Rohr e zur Lampe, bis
die Flüssigkeit den Luftzutritt durch das Rohr g
abschlieſst.
Tafeln