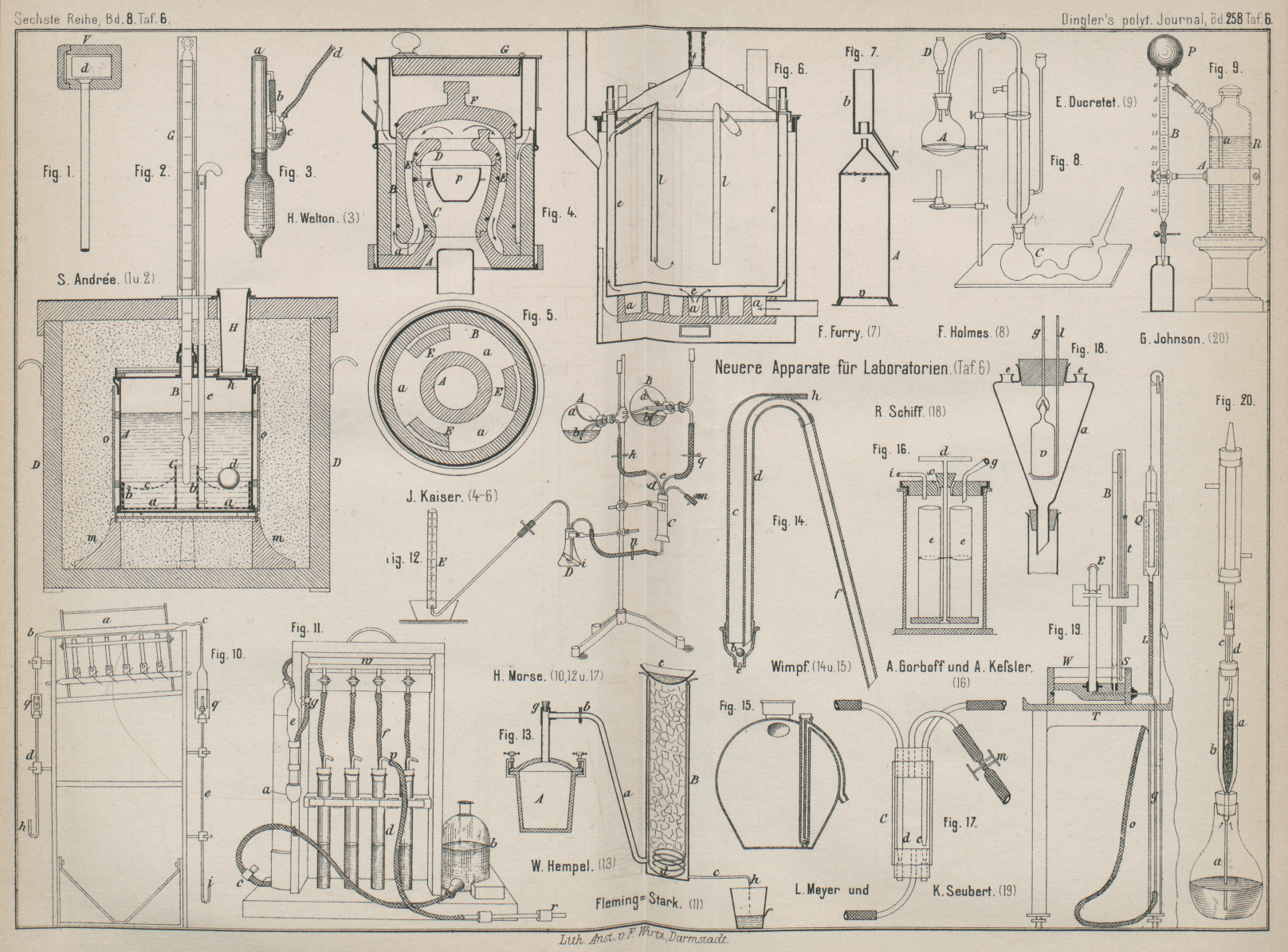
| Titel: | Neuere Apparate und Verfahren für chemische Laboratorien. |
| Fundstelle: | Band 258, Jahrgang 1885, S. 72 |
| Download: | XML |
Neuere Apparate und Verfahren für chemische
Laboratorien.
(Fortsetzung des Berichtes Bd. 254 S.
67.)
Mit Abbildungen im Texte und auf Tafel 6.
Neuere Apparate und Verfahren für chemische
Laboratorien.
S. A. Andrée beschreibt im Jernkontorets Annaler 1884 bezieh. im Stahl und
Eisen, 1885 S. 144 ein Hydropyrometer für Zwecke
der Metallurgie. In dem Gefäſse A (Fig. 1 und 2 Taf. 6) aus
dünnem Messingblech befindet sich der Mischer C,
welcher aus einem Siebbleche a und zwei gelochten
Ringen b besteht, zwischen denen ein Drahtgewebe c befestigt ist, um den erhitzten Eisencylinder d aufzunehmen, worauf der Rührer durch den aus
Fischbein hergestellten Stab e auf- und abbewegt wird.
Beim Hineinwerfen des Probecylinders d durch das Rohr
H öffnet sich die Klappe h, um sofort durch ein Gegengewicht wieder geschlossen zu werden. Das Wassergefäſs ist auf
4 Klötzen m befestigt und mit Pappdeckel o umgeben; der übrige Raum des Gefäſses D ist mit loser Baumwolle gefüllt. Ein geschlitztes
Metallrohr G soll das Thermometer schützen. Die
Erhitzung des Eisencylinders d geschieht in bekannter
Weise in der in Fig.
1 gezeichneten eisernen Kapsel V. (Vgl. F. Fischer 1877 225 *
468.)
H. Landolt beschreibt in den Berichten der deutschen chemischen Gesellschaft, 1885 * S. 56 eine Vereinigung von Heiſswassertrichter mit Wasserbad (Textfig. 1). Ein aus Kupferblech angefertigter Kasten
ist hinten 11cm, vorn 9cm hoch. In die als Wasserbad dienende höhere
Abtheilung lassen sich durch 3 Oeffnungen Bechergläser u.s.w. einsenken und es
kommen diese auf eine durchlöcherte Blech platte zu stehen, welche im Inneren des
Kastens 1cm hoch über dem Boden sich befindet. Die
vordere niedrigere Abtheilung ist von drei oben mit vorstehendem Rande versehenen
Blechtrichtern durchsetzt, in welche die Glastrichter gesteckt werden. Der Apparat
steht auf vier 20cm hohen Füſsen, von denen einer
mit Stellschraube versehen ist. Um die Flamme des Gasbrenners, mit welcher man den
hinteren Theil erhitzt, von den unter den Trichtern stehenden Gläsern abzuhalten,
ist am Boden des Kastens ein senkrechtes Schutzblech angebracht. Zum Auswaschen der
Niederschläge dient eine stets im Wasserbade stehende Spritzflasche, welche auf die
in der Figur ersichtliche Weise mit Kautschukröhren versehen ist.
Fig. 1., Bd. 258, S. 73J. A. KaiserWissenschaftliche Beilage zum Programm der St.
Galler Kantonschule, 1882. (Gef. eingeschickt.)
beschreibt einen Glühofen für Gasfeuerung. Die
Chamottestücke A bis D
(Fig. 4
und 5 Taf. 6)
ruhen auf einer von 3 Füſsen getragenen Blechplatte, während der Ring E nur an 3 Stellen die Platte A berührt, somit 3 Oeffnungen a für den
Durchgang der Verbrennungsgase frei läſst. Nach dem Abheben des Deckels G können die Stücke F und
D entfernt werden, um den Tiegel p in das Platindreieck e
zu setzen (vgl. F. Fischer im Jahresbericht der chemischen Technologie, 1884 * S. 1319).
Unter dem doppelwandigen Trockenschranke (Fig. 6 Taf. 6)
befindet sich ein. eiserner Kasten, dessen ringförmige Zwischenräume a von der seitlich eintretenden Luft durchzogen werden,
so daſs sie heiſs in den Mantel e tritt, um durch Rohre
l auf die Oberfläche der zu verdunstenden
Flüssigkeit oder in die zu trocknende Flasche eingeführt zu werden.
V. Meyer2 V. Meyer: Pyrochemische Untersuchungen
(Braunschweig 1885. Friedr. Vieweg und
Sohn.) fand bei seinen pyrochemischen Untersuchungen, daſs Schmiedeisen und bestes Porzellan leichter
schmelzen als Platin. Sauerstoff und Stickstoff haben bis 1700° dieselbe Ausdehnung.
Kohlenoxyd zerfällt bei 1700° bereits theilweise
nach der Formel 2CO = CO2 + C unter Abscheidung von
Kohlenstoff. Kohlensäure zeigt in einem Platinapparate
selbst bei 1700° noch keine merkbare Zersetzung. Durch ein mit Porzellanbrocken
gefülltes Porzellanrohr geleitet, beginnt sie aber bereits bei 1300° theilweise in
Kohlenoxyd und Sauerstoff zu zerfallen, so daſs diese Erscheinung durch rauhe
Flächen befördert wird.
Stickoxyd bleibt bei 1200° unverändert, zerfällt aber
bei 1700° vollständig in Sauerstoff und Stickstoff. Salzsäure zerfällt theilweise in Chlor und Wasserstoff. Wasserdampf beginnt schon bei 1200° in seine
Bestandtheile zu zerfallen, so daſs von diesen Verbindungen die Kohlensäure am
beständigsten ist.
Beschädigte Platinapparate können dadurch ausgebessert werden, daſs man die zu löthende Stelle
mit einer Gasflamme rothglühend macht, ein Platinblech auflegt und nun eine
Knallgasflamme darauf richtet, bis das Blech schmilzt. Sobald das Platin anfängt,
Funken zu sprühen, wobei die Kugel zerflieſst und die Oeffnung bedeckt, muſs man die
Flamme entfernen. Nach dem Abkühlen entfernt man die hervorragenden Theile mit einer
Schlichtfeile und glättet mit Schmirgelpapier.
J. W. Pratt (Chemical News, 1885 Bd. 51 S. 181) legt auf
die beschädigte Stelle von Platingeräthen gepulvertes
Goldchlorid und erhitzt allmählich, schlieſslich mit dem Gasgebläse, bis das Gold
geschmolzen ist.
H. Welton (Génie civil, 1885 Bd. 6 * S. 257) verbindet
die zum Messen von Brom, Säuren u. dgl. bestimmte Pipette (Fig. 3 Taf. 6) durch ein
seitliches Rohr und den Schlauch b mit einer kleinen
Waschflasche e, welche Natronlauge enthält. Man
schlieſst die Oeffnung a mit dem Finger und saugt am
Schlauche d.
Den Wimpf'schen Heber
liefert die Möncheberger Gewerkschaft in Kassel aus
Thon in der durch Fig. 14 Taf. 6 veranschaulichten Form. Der kürzere Schenkel d ist von einer weiteren Röhre c umgeben, welche oben geschlossen und am unteren Ende e mit Kugel- oder Kegelventil b versehen ist. Beim Eintauchen des Apparates in die abziehende
Flüssigkeit hebt sich das Ventil und das Rohr c sowie
der Schenkel d füllen sich beide gleichmäſsig bis zur
Höhe der abzuziehenden Flüssigkeit. Bläst man nun durch den oben bei h angebrachten Rohransatz Luft ein, so wird die
Flüssigkeit, da die Oeffnung e geschlossen ist, durch
d nach f getrieben und
der Heber tritt in Wirksamkeit, worauf man sofort mit Blasen aufhört und die
Anblaseöffnung auf irgend eine Weise verschlieſst. Jetzt lastet kein Druck auf dem
Ventile b, die Flüssigkeit kann ungehindert
nachflieſsen und das Gefäſs bis auf den letzten Rest entleert werden. Will man das
Abhebern unterbrechen, so ist es nur nöthig, wiederum, aber diesmal etwas
anhaltender, in h zu blasen, wodurch das Ventil b geschlossen wird und der Heber abläuft. Soll das
Abziehen wieder beginnen,
so hat man abermals in das Rohr h zu blasen, so daſs
das Spiel von Neuem beginnt. Vortheilhaft ist es ferner, die für Salpetersäure
bestimmten Thongefäſse mit derartigen Hebern zu verbinden, wie Fig. 15 Taf. 6 zeigt.
E. Ducretet (Revue industrielle, 1884 * S. 491)
befestigt nach Fig.
9 Taf. 6 die Bürette B durch eine Spange A mit der Flasche R,
welche die für Alkalimetrie oder Hydrotimetrie bestimmte Maſsflüssigkeit enthält.
Preſst man den Gummiballen P zusammen und verschlieſst
beim Wiederaufblähen die obere Oeffnung desselben mit dem Finger, so wird die
Bürette durch das Rohr a in bekannter Weise
gefüllt.
Um im luftverdünnten Raume bei erhöhter Temperatur trocknen zu können, hängt R.
Anschütz (Liebig's Annalen der Chemie, 1885 Bd. 228 * S. 305) in die
Luftpumpenglocke ein in der Ebene spiralförmig gerolltes Bleirohr, dessen Enden nach
auſsen führen, um Wasserdampf hindurchleiten zu können. Die flache Spirale ist
beiderseitig mit Nickeldrahtnetz überzogen, so daſs eine für Tiegel u. dgl.
geeignete Trogvorrichtung entsteht.
H. N. Morse verwendet nach dem American Chemical Journal, 1885 * S. 60 zur Reinigung von Quecksilber durch Destillation ein etwa 45cm langes, an beiden Enden ausgezogenes
Verbrennungsrohr a (Fig. 10 Taf. 6), welches
in einer durch Gasflammen erhitzten schmiedeisernen Rinne liegt. Das Ende b ist durch einen Schlauch mit dem Rohre d, das Ende c durch
Gummistopfen mit dem 83cm langen Rohre e verbunden, welches etwas in c hineinragt, damit der Stopfen nicht mit heiſsem Quecksilber in Berührung
kommt. Beide Verbindungen sind mit Quecksilberzellen q
umgeben. Die aufwärts gebogenen Enden h und i sind 6cm lang, um
die Schwankungen des Atmosphärendruckes auszugleichen; die Entfernung von b bis h beträgt etwa 77cm. Man taucht nun das Ende h in einen mit dem zu destillirenden Quecksilber
gefüllten Cylinder, verbindet das Ende i mit einer
Quecksilberluftpumpe, so daſs das Quecksilber in das Rohr a steigt und die Destillation beginnt. Sobald das Rohr e mit dem destillirten Quecksilber bis zur
Barometerhöhe gefüllt ist, entfernt man die Pumpe, worauf der Apparat selbstthätig
weiter arbeitet.
Zur Werthbestimmung des Zinkstaubes verwendet Morse (daselbst * S. 52) zwei Literflaschen A und B (Fig. 12 Taf. 6), deren
doppelt durchbohrte Stopfen zwei Glasröhren a und b tragen. Die eine Flasche ist etwa zu ⅓ mit Wasser,
die andere mit ebenso viel Salzsäure gefüllt; die Flüssigkeiten werden zum Sieden
erhitzt, so daſs die Luft durch die Rohre a entweicht.
Um den Zufluſs aus den Flaschen in das 7cm lange
Rohr C (vgl. 17 Taf. 6) zu unterbrechen, schiebt man
die Rohre d und e so tief,
daſs die Spitzen in die Bohrungen des unteren Stopfens treten, welche durch
Glasstäbe geschlossen sind. Hebt man Rohr d, öffnet
Quetschhahn k und saugt am Schlauche m, so füllt sich das Rohr C
aus der Flasche A. Man wiegt in das Rohr i
etwa 0g,2 Zinkstaub ab, fügt ausgekochtes Wasser
hinzu, mischt und schiebt einen mit Wasser getränkten Stopfen aus Glaswolle bis auf
das Gemisch. Man bringt nun das Rohr i in der Flasche
D in die Lage Fig. 12 und läſst durch
Oeffnen der Quetschhähne k, q und n Wasser sowie Salzsäure eintreten und erwärmt die
Flasche. Der entwickelte Wasserstoff wird im Meſsrohre E aufgefangen.
J. Fleming-Stark (Journal of the Society of Chemical
Industry, 1885 * S. 311) verwendet zur Bestimmung
des Chlor es in den aus Chlorkalkkammern entweichenden Gasen eine Bürette
a (Fig. 11 Taf. 6), welche
mit der Wasserflasche b durch einen Schlauch verbunden
ist. Der Hahn c hat eine grobe und eine feine Bohrung:
erstere wird geöffnet, wenn durch Heben der Flasche b
die in der Bürette a enthaltene Luft durch den zwischen
der Bürette und der Waschflasche e befindlichen
Zweiwegehahn g ausgetrieben werden soll, letztere, wenn
bei gesenkter Flasche b durch das in die
Chlorkalkkammer ragende Rohr r Gase angesaugt werden.
Diese gehen durch die bis auf den Boden der halb mit Jodkaliumlösung gefüllten
Glascylinder d reichenden Rohre v, dann durch Schlauch f und Hahnrohr w in die Jodkalium und Stärkekleister enthaltende
Flasche e und durch Hahn g
in die Bürette a. Sind etwa 400cc Gas durch die Jodkaliumlösung im Rohre d durchgesaugt, so wird diese in bekannter Weise mit
Arsenigsäure titrirt.
Um Niederschläge von dem Filter zu trennen, empfiehlt
F. A. Gooch in der Chemical
News, 1885 Bd. 51 S. 230 statt Papierfilter mit Alkohol befeuchtetes Anthracen als Filtermittel zu verwenden, welches nach
beendigter Filtration durch Benzol o. dgl. gelöst wird.
R. Schiff (Berichte der deutschen chemischen
Gesellschaft, 1885 S. 1538) befestigt zur Bestimmung des specifischen Gewichtes von Flüssigkeiten bei höherer
Temperatur ein birnenförmiges Gefäſs a (Fig. 18 Taf.
6) mittels Stopfen auf einem gewöhnlichen Rundkölbchen, in welchem je nach Bedarf
die verschiedenen zur Erwärmung des Apparates geeigneten Flüssigkeiten zum Sieden
erhitzt werden. Von den beiden engeren Rohransätzen e
des Gefäſses steht der eine mit einem Rückfluſskühler in Verbindung, während in dem
anderen ein Thermometer eingesetzt ist. In der mittleren weiten Oeffnung sitzt ein
doppelt durchbohrter Kork, in dessen eine Durchbohrung ein mit einem eigenthümlich
geformten Helme in Verbindung stehendes Glasröhrchen g
eingesetzt ist, während durch die andere der Stiel eines eisernen Löffelchens l geht. Auf l ist (mit ein
wenig Papierunterlage) das eigentliche Pyknometer v
eingesetzt, dessen Hals in einer capillaren Spitze endet, die jedoch während der
Wägungen mit einem dünnen Glaszäpfchen verschlieſsbar ist. Der Hals hat eine
kegelförmige, nach oben spitz zulaufende Erweiterung, auf welche der erwähnte Helm
luftdicht aufgeschliffen ist. Durch Auf- und Niederdrücken des eisernen Löffelchens
kann man das Pyknometer leicht in den Helm einsetzen oder daraus entfernen. Somit ist das
Innere des Fläschchens sammt Helm gegen die äuſseren Dämpfe völlig abgeschlossen.
Beim Gebrauche des Apparates läuft die Flüssigkeit so lange aus der Spitze in den
Helm, bis die Temperatur der äuſseren Dämpfe genau erreicht ist, was nach etwa 10
Minuten stets eintreten wird- hierbei ist die austretende Flüssigkeitsmenge nicht
verloren, sondern wird in der Biegung des Helmes wiedergefunden. Nach beendigter
Erhitzung hebt man den Apparat beim Stopfen aus dem Mantel, nimmt das Pyknometer ab,
verschlieſst es und wägt, nachdem es erkaltet ist. Hierauf setzt man den ganzen
Apparat wieder ein und ist bereit, mittels einer höher siedenden Heizflüssigkeit
eine weitere Bestimmung zu machen.
A. Gorboff und A. Keſsler
(daselbst * S. 1363) verwenden, um bei fractionirter
Destillation unter vermindertem Drucke die Vorlagen leicht wechseln zu
können, einen Glascylinder, dessen mit Paraffin getränkter und mit Kautschukring
umspannter Kork das zur Luftpumpe führende Rohr i (Fig. 16 Taf.
6) und das vom Siedegefäſse kommende Rohr g trägt. In
der mittleren Oeffnung steckt ein kurzes Glasrohr, das durch Kautschukstopfen c geschlossen wird, in welchen sich der Glasstab d leicht drehen und verschieben läſst, um die Vorlage
e bequem wechseln zu können, ohne die
Luftverdünnung aufzuheben.
Fig. 2., Bd. 258, S. 77Fig. 3., Bd. 258, S. 77W. Hempel (daselbst * S. 1434) verwendet als
Filterpresse für Laboratorien durchlochte
Porzellanplatten a (Textfig.
2 und 3) mit dazwischen liegendem
Gummiringe b; in letzterem ist in ein seitliches Loch
eine Glasröhre c eingeschoben, an welche sich die etwa
3m lange Druckleitung C anschlieſst. Als Träger der Filter A dient
ein eisernes Gestell B mit Glastafeln d und Glasrinne e. Soll
der Apparat benutzt werden, so nimmt man die Filter aus einander, legt auf die
Porzellanplatten a zuerst ein grobes, passend
geschnittenes Stück Leinwand, dann ein Stück Flieſspapier, hierauf den Gummiring b, dann wieder ein Stück Flieſspapier, ein zweites
Leintuch und endlich die zweite Porzellanplatte. Das Ganze preſst man mit 4 eisernen
Schrauben, über welche Gummischuhe gezogen sind, zusammen. Die zu filtrirende
Flüssigkeit wird in den Trichter f gegossen.
Der Apparat zur Herstellung von Fluorwasserstoffsäure
und Kieselfluorwasserstoffsäure
besteht aus einem
guſseisernen Topfe A (Fig. 13 Taf. 6), in
dessen aufgeschraubtem Deckel eine eiserne Gasleitungsröhre eingesetzt ist, welche
bei b mit einem Bleirohre a verbunden wird. Die Oeffnung g kann durch
einen Kork verschlossen werden. Der Absorptionsapparat B besteht aus einem einfachen, etwa 15cm
weiten und 70cm hohen Bleicylinder, welcher am
Boden ein etwa 1cm weites Ablaufrohr c hat. In dem Bleicylinder liegt eine Kühlschlange d, welche aus einem etwa 5mm weiten Bleirohre zusammengerollt ist; aller übrige Raum ist mit groben
Holzkohlenstücken ausgefüllt.
Beim Gebrauche beschickt man den eisernen Topf mit 1k gemahlenem Fluſsspath und 1k
concentrirter roher Schwefelsäure, rührt die Masse gut um und erhitzt am besten über
freiem Feuer. Man gieſst dann in eine auf den Absorptionscylinder gestellte
Bleischale e, in deren Boden einige Löcher gestochen
sind, etwa 750cc Wasser und stellt bei f ein Gummi- oder Bleigefäſs unter. Durch die
Kühlschlange läſst man einen starken Strom kalten Wassers laufen. Das Wasser
vertheilt sich aus der Bleischale auf die Holzkohlen und flieſst über diese dem
Fluorwasserstoffgase entgegen; die gebildete Fluſssäure sammelt sich in dem Gefäſse
f. Hört das Tropfen bei h auf, so gieſst man die abgelaufene Fluſssäure wieder zurück nach e und wiederholt dies so lange, bis der eiserne Topf
zum schwachen Rothglühen gebracht ist. Nach dem Erkalten findet man beim Oeffnen des
eisernen Topfes eine kaum noch sauer reagirende, trockene Masse von Gyps, welche
sich mit einem eisernen Meiſsel sehr leicht herausstechen läſst.
Aus dieser rohen Fluorwasserstoffsäure erhält man dadurch reines Fluorammonium, daſs man sie in 2 Hälften theilt, den einen Theil
mit Ammoniak bis zur alkalischen Reaction absättigt, dann wieder mit dem anderen
Theile zusammengieſst und hierauf in einer Platinschale eindampft. Es scheidet sich
beim Concentriren Fluorwasserstoff-Fluorammon ab; ist dies erreicht, so läſst man
erkalten und trennt die Krystalle von der Mutterlauge durch Filtration in einem mit
Wachs überzogenen Glas- oder einem Guttaperchatrichter. Durch zweimaliges
Umkrystallisiren gelingt es leicht, dieses Salz chemisch rein herzustellen, welches
sich in einem Holz- oder Papierkasten ohne Veränderung aufbewahren läſst.
Um Kieselfluorwasserstoffsäure darzustellen, befestigt
man an die Flansche b mittels eines Korkes eine
rechtwinkelig gebogene weite Glasröhre. Ist die Glasröhre wenigstens 18mm im Lichten weit, so ist es nicht nöthig, das
entwickelte Fluorsilicum unter Quecksilber austreten zu lassen, da der Gasstrom
leicht im Stande ist, das ausgeschiedene Kieselsäurehydrat heraus zu treiben, auch
wenn man die Glasröhre unmittelbar in Wasser tauchen läſst.
G. St. Johnson (Chemical News, 1885 Bd. 52 * S. 39)
bringt die Stoffe, welche mit flüchtigen Lösungsmitteln
ausgezogen werden sollen, in das oben erweiterte Rohr a (Fig. 20 Taf. 6). Die in
der Kochflasche entwickelten Dämpfe steigen in dem weiteren Rohre b auf, gelangen durch Rohr c zum Kühler, das verflüssigte Lösungsmittel tropft durch Rohr d auf die Probe im Rohre a, die gebildete Lösung flieſst in die Kochflasche zurück.
Zur Werthbestimmung des schwefelsauren Ammoniums bringt
F. G. Holmes (daselbst * S. 49) 0g,5 der Probe mit 20cc Wasser in die Flasche A (Fig. 8 Taf. 6), 25cc Zehntel-Normalschwefelsäure in den Kugelapparat
C, läſst durch das Tropfgefäſs D 15cc einer 20
procentigen Natronlauge einflieſsen und destillirt. Schlieſslich wird Luft durch den
Apparat gesaugt und die nicht gesättigte Schwefelsäure zurücktitrirt.
Zur Bestimmung des Feuchtigkeitsgehaltes von
Futtermittel verwendet F. E. Furry (daselbst
1884 Bd. 50 * 8. 293) einen 18cm langen, 7cm weiten Zinncylinder A (Fig.
7 Taf. 6) mit Siebboden s. Derselbe wird
umgekehrt, mit der Probe gefüllt, der Siebboden v
aufgelegt, der Cylinder wieder aufrecht gestellt und das Abzugsrohr b aufgesetzt. Nun wird erwärmt und das im Rohre b verflüssigte Wasser durch Rohr r abgelassen, schlieſslich wieder gewogen.
Zur Bestimmung des Kohlenstoffes in Eisen und Stahl
behandelt Th. Turner nach dem Iron, 1885 Bd. 26 * S. 84 die Probe mit dem aus 53,4 Th. Chlorammonium und
85,4 Th. krystallisirtem Kupferchlorid bestehenden Gemisch, von welchem 15g in 50cc Wasser
für 1g Eisen ausreichen. Billiger ist eine Lösung
von 360g krystallisirtem Kupfersulfat in 750cc Wasser unter allmählichem Zusätze von 310g Chlornatrium und Abfiltriren des ausgeschiedenen
Natriumsulfates. Zur Sammlung des Kohlenstoffes dient ein ausgezogenes
Verbrennungsrohr (Textfigur 4), in welches zunächst
eine Thonkugel a, dann ausgeglühter Sand, eine Lage
Asbest und nochmals Sand gebracht wird. Nach dem Filtriren wird ausgewaschen,
getrocknet, das Rohr in einen einfachen Verbrennungsofen gelegt und der Kohlenstoff
in bekannter Weise im Sauerstoffstrome verbrannt. Die gebildete Kohlensäure wird in
einem Kaliapparate aufgefangen und gewogen.
Fig. 4., Bd. 258, S. 79Bei der Untersuchung Kohlenstoff reicher Gase
mäſsigen L. Meyer und K.
Seubert nach Liebig's Annalen, 1885 Bd. 226 *
S. 87 die Explosion durch stark verminderten Druck, ohne mit Luft zu verdünnen.Vgl. Ferd. Fischer: Chemische Technologie der
Brennstoffe, S. 238. In die mit guſseisernem Boden
versehene Quecksilberwanne W (Fig. 19 Taf. 6) ist
mittels einer Eisenfassung das Barometerrohr B und
mittels eines eingedrehten eisernen Zapfens das etwa 40cm lange Eudiometerrohr E eingesetzt. Beide
stehen in Verbindung mit einer wagerechten Bohrung im Boden der Wanne, die
auſserhalb der Wanne als eiserner Ansatz endigt, in welchen das Glasrohr g luftdicht eingekittet ist. Dasselbe biegt sich bald
abwärts und reicht, die Platte des Tisches T
durchsetzend, bis nahe zum Boden herab. Ein am unteren Ende von g befestigter, stark übersponnener Kautschukschlauch o vermittelt die Verbindung mit dem Quecksilberbehälter
Q, welcher in einem hölzernen Schlitten in den
Laufleisten L auf und ab bewegt werden kann. Man
entfernt das Eudiometer E und läſst durch Senken von
Q das Quecksilber aus der Wanne abflieſsen, bis das
Capillarrohr des Barometers nicht mehr eintaucht. Schiebt man nun unter die untere
Mündung des Capillarrohres ein passendes Gefäſs mit Wasser und setzt jetzt das mit
Quecksilber gefüllte Eudiometer wieder ein, so kann man durch Heben von Q die Luft aus dem Barometer austreiben und durch
Senken Wasser wieder einziehen. Indem man Luft nachströmen läſst, zieht man das
Wasser abwärts bis unter die Spitze S, neben welcher
sich, wenn das Quecksilber wieder gehoben wird, ein Theil des Wassers ansammelt,
während der Ueberschuſs mit der Luft durch das jetzt wieder in das Quecksilber der
Wanne tauchende Capillarrohr hinausgedrückt wird. Durch wiederholtes Heben und
Senken von Q läſst sich die Luft so gut wie vollständig
aus B verdrängen. Die Wände des Rohres bleiben dabei
genügend benetzt, um die der jedesmaligen Beobachtungstemperatur entsprechende
Tension des Wasserdampfes zu liefern.
Die Füllung des in der Kuppe mit einem Wassertröpfchen befeuchteten Eudiometers
geschieht nach dem üblichen Bunsen'schen Verfahren.
Nachdem das mit Quecksilber gefüllte Rohr in den Boden der Wanne eingesetzt ist,
vermindert man zunächst den Druck möglichst, um etwa am Glase haftende Luftbläschen
loszulösen und durch Heben von Q nach oben zu treiben.
Sollte sich an den Drähten des Eudiometers eine Luftblase zeigen, so wird dieselbe,
nachdem das Quecksilber in Q mit dem Stande der Wanne
gleich gestellt worden, durch Ausheben und Umkehren des Rohres in gewohnter Weise
entfernt.
Mit der erforderlichen Menge Sauerstoff gemischt, verbrennen die Gase beim
Durchschlagen eines schwachen Inductionsfunkens bei folgendem Drucke: Methan bei
130mm, Propan 71mm, Aethylen und Propylen 63mm, Acetylen
32mm, Kohlenoxyd 219mm und Wasserstoff bei 125mm.
Nachdem das Eudiometer in angegebener Weise völlig mit Quecksilber
gefüllt ist, wird das zu analysirende Gas eingeführt; die Menge desselben ist so zu
bemessen, daſs nach dem Zusätze des erforderlichen Sauerstoffes das Eudiometer bei
1at Druck nur etwa zu 0,1 gefüllt sein würde.
Ist z.B. die lichte Weite des Rohres 18mm, sein
Querschnitt demnach 2qc,5 und bei einer Länge von
40cm sein gesammter Inhalt 100cc, so sind etwa 10cc des explosiven Gemisches anzuwenden. Bei Gasen indessen, welche zur
Explosion einer weniger starken Druckverminderung bedürfen, kann über die angegebene
Grenze entsprechend hinausgegangen werden.
Um die Abmessung der kleinen Gasvolumen zu erleichtern, verwendet
man bei der Luftkalibrirung des Eudiometers ein kleines Maſsgefäſs von kaum 2cc Inhalt und nimmt von den dichteren Gasen etwa
1, von den leichteren 2 bis 3 Vol. zur Analyse. Es empfiehlt sich beim Ablesen, das
Gefäſs Q so einzustellen, daſs die Kuppe des
Quecksilbers in B mit dem zugeschmolzenen Ende des
Eudiometers E etwa in gleicher Höhe steht.
Die Beleuchtung der Skalen geschieht durch einen an E angebrachten weiſsen Papierschirm. Es gelingt auf
diese Weise leicht, die Ablesungen bis auf 0mm,1 abzuschätzen.
Der Stand der Quecksilberkuppe in B und die von dem am
Barometerrohre hängenden Thermometer t angezeigte
Temperatur werden wie gewöhnlich abgelesen.
Man vermindert nunmehr den Druck auf die oben angegebenen Werthe
und läſst den Funken überschlagen. Sollte derselbe die Explosion nicht bewirken, so
kann durch Heben von Q der Druck in kürzester Zeit in
erforderlichem Maſse gesteigert werden. Die Explosion erfolgte stets mit schönem
Licht und so ruhig, daſs trotz der geringen entgegenwirkenden Quecksilbersäule nur
ein schwaches Schwanken der Quecksilberkuppe in E
eintrat und niemals Gasblasen bis zum unteren Ende des Eudiometers geschleudert
wurden. Durch Senken und Heben von Q bespült man die
Eudiometerwände mit Quecksilber und stellt schlieſslich zur Ablesung Druck und
Volumen wieder annähernd gleich ein.
Zur Absorption der Kohlensäure bedient man sich zweckmäſsig der
von Bunsen empfohlenen Natronlauge von 7 Proc. NaOH.
Eine genügende Menge derselben wird unmittelbar vor der Verwendung in einem Erlenmeyer'schen Becher etwa 10 Minuten lang gekocht,
um die absorbirte Luft auszutreiben und sodann noch warm in das Eudiometer
eingeführt. Es geschieht dies, nachdem man das Quecksilber in Q und in der Wanne auf gleiche Höhe gebracht und E ans der Bohrung herausgehoben hat, in gewohnter Weise
mittels einer Hakenpipette. Sobald das Eudiometer wieder eingesetzt ist, wird der
Druck so geregelt, daſs die Lauge möglichst nahe an die Eudiometerdrähte
hinaufsteigt, ohne dieselben jedoch zu erreichen. Sollte letzteres eintreten, so
entsteht ein freilich meist ganz unbedeutender Fehler dadurch, daſs sich Lauge
zwischen Glas und Drähten in die Höhe zieht, dort hängen bleibt und so das Volumen
des Gases etwas zu groſs finden läſst Man kann sich übrigens durch Messung des
Volumens der Lauge vor dem Hinauftreiben derselben auch vor diesem geringfügigen
Fehler schützen.
Die Absorption der Kohlensäure geht in dem kleinen Gasvolumen sehr
rasch vor sich und kann schon nach ½ Stunde als beendet angesehen werden. Man stellt
wie früher Druck und Volumen annähernd gleich ein und liest ¼ Stunde später ab,
wobei diesmal natürlich auch der Stand der Lauge beobachtet wird.
Zur Berechnung der Analysen werden
die an der Theilung des Eudiometers abgelesenen Volumen wie gewöhnlich nach der
Kalibrirungstabelle berichtigt. Der Druck des Gases ist gleich dem Unterschiede
zwischen der in gleicher Höhe mit der Quecksilberkuppe in E auf der Skala von B abgelesenen Zahl und
der an der Kuppe des Quecksilbers in B abgelesenen.
Eine Richtigstellung für die Tension des Wasserdampfes ist nicht anzubringen, weil
letztere im Barometerrohre gleichfalls herrscht. Als einzige Berichtigung des
Druckes ist die Reduction der an der Glasskala abgelesenen Länge der
Quecksilbersäule auf 0° anzubringen, welche bei der meist sehr geringen Gröſse
derselben kaum ins Gewicht fällt. Bei der Absorption der Kohlensäure ist noch die
der Höhe der Natronlauge im Rohre entsprechende Quecksilbersäule vom Drucke
abzuziehen, dagegen die Abweichung zwischen der Dampftension über reinem Wasser und
jener über der Natronlauge dem Drucke hinzuzufügen.
Fr. Stolba empfiehlt in den Sitzungsberichten der böhmischen Gesellschaft der Wissenschaften, 30.
Januar 1885, die Verwendung von Nickelapparaten für
chemische Laboratorien. Schalen und Tiegel aus Nickelblech rosten nicht und sind bei
Glühhitze sehr beständig; sie dürfen aber nicht in unmittelbare Berührung mit
glühender Kohle kommen, da sie dann brüchig werden. Die beste Art der Erhitzung ist
jene mit der Gaslampe oder dem Gasofen; allein auch hierbei ergibt sich ein
eigenthümlicher Uebelstand. Selbst aus solchen Flammen, welche nicht im geringsten
ruſsen und mit nicht leuchtender Flamme brennen, scheidet sich am Nickelmetalle eine
reichliche Rufsschicht ab, welche fortwährend an Stärke zunimmt und schlieſslich abfällt. Obgleich man
etwas ähnliches auch bei anderen Metallen beobachten kann, findet dies bei keinem
anderen in so auffallendem und unangenehmen Grade statt und muſs man, um den
Uebelstand möglichst zu mindern, Flammen anwenden, denen man die gröſste zulässige
Luftmenge zuführt. Die Nickeltiegel selbst leiden durch diese Ruſsschichte nicht;
nur werden sie dadurch an der unteren Seite verunreinigt und büſsen daselbst ihr
schönes Ansehen ein. Nach dem Gebrauche werden sie am besten zunächst mittels einer
Drahtbürste oder mittels feiner Eisensiebe und schlieſslich mit Seesand
gereinigt.
Die Nickelschalen eignen sich sehr gut zum Ausglühen und Veraschen mancher Stoffe,
zur Behandlung anorganischer und organischer Präparate mit Aetzlaugen und
kohlensauren Laugen, insbesondere sehr gut zum Schmelzen mit salpetersauren Alkalien
und Aetzalkalien, da sie hierbei nur sehr unbedeutend angegriffen werden, und zu
demselben Zwecke mit bestem Erfolge sehr oft verwendet werden können. Sehr angenehm
ist bei diesen Arbeiten der Umstand, daſs das Nickel so schwer schmilzt, nämlich
erst in starker Weiſsglut, so daſs man nicht so bald in die Lage kommen wird, eine
Schmelzung der Schale befürchten zu müssen. Das Nickelblech wird von den meisten
anorganischen und organischen Säuren auch bei starker Verdünnung derselben mehr oder
weniger angegriffen, namentlich bei Luftzutritt und längerer Einwirkung. Dasselbe
gilt auch von sauer reagirenden Salzlösungen, z.B. von der Lösung des Alauns,
Weinsteins o. dgl. Hieraus folgt, daſs man solche Stoffe von den Nickelgeräthen fern
halten muſs. Dagegen widersteht es in bemerkenswerthem Grade der Einwirkung der
concentrirten Schwefelsäure, so daſs man manche Zersetzungen von Mineralien mittels
der genannten Säure ganz gut in Nickelgeräthen vornehmen kann. Auch von Blei und
Bleioxyd wird Nickel nicht angegriffen.
Nickel eignet sich ferner sehr gut zur Herstellung von Federzangen (sogen.
Pincetten), Spateln, Tiegelzangen u. dgl.
Tafeln