| Titel: | Neuerungen im Metallhüttenwesen. |
| Fundstelle: | Band 271, Jahrgang 1889, S. 214 |
| Download: | XML |
Neuerungen im Metallhüttenwesen.
(Fortsetzung des Berichtes S. 172 d. Bd.)
Mit Abbildungen auf Tafel
11.
Neuerungen im Metallhüttenwesen.
Kupfer und Phosphorkupfer. Um Kupfererze auf nassem Wege
zu verarbeiten, müssen dieselben entweder oxydisch sein oder, falls geschwefelte
Erze verwendet werden sollen, in den oxydirten Zustand übergeführt werden, was in
der Regel durch Röstung geschieht. Joseph Perino
schlägt nun ein Verfahren vor, um womöglich gänzlich ohne Röstung den Kupferkies,
das hervorragendste Kupfererz, zu sulfatisiren. Dasselbe besitzt nach seiner
Zusammensetzung als Mineral die Formel CuFeS2 und
enthält demnach 34,57 Proc. Kupfer und 30,54 Proc. Eisen. Selten aber tritt der
Kupferkies ganz rein auf, sondern ist in der Regel vergesellschaftet mit
Schwefelkies (FeS2), ja in den meisten Fällen hat es
der Hüttenmann nur mit Kupferkies haltigen Schwefelkiesen zu thun. Der trockene Weg
mit seiner Bildung von Stein und dem wiederholten Abrösten und Einschmelzen ist
sehr umständlich, weshalb das Auflösen geschwefelter Kupfererze mittels
Eisenchlorides oder Eisenoxydsulfates erst neuerdings wieder von Siemens und Halske (1888 269
364) behufs Gewinnung eines geeigneten Elektrolyten für elektrolytische
Kupfergewinnung vorgeschlagen ist. Perino benutzt nun
salpetersaure Eisensalze (Eisennitrate), um das Schwefelkupfer der Erze bei niederen
Temperaturen (50 bis 150°) direkt zu schwefelsaurem Salze (Kupfersulfat) zu
oxydiren. Das stets vorhandene Schwefeleisen soll sich an dieser Oxydation nicht
betheiligen, wenn der Prozeſs richtig geleitet wird. Perino beschreibt in der Berg- und
Hüttenmännischen Zeitung, 1888 S. 177 ff., selbst die von ihm zu dem
angegebenen Zwecke gemachten Versuche.
Mischt man nämlich nach Angabe Perino's gemahlenen
Kupferkies innig mit Eisennitrat und setzt das Gemenge einer Temperatur von etwas
über 40° aus, so beginnt schon die Entwickelung der nitrosen Dämpfe unter Bildung
von Kupfersulfat. Steigert man die Temperatur allmählich bis 100° und 150°, so
erreicht man es, daſs beim nachherigen Auslaugen mit Wasser eine reine
Kupfervitriollauge resultirt, ohne eine Spur von Eisengehalt, während im Rückstande
sich unzersetztes Schwefeleisen, Schwefelsilber u.s.w. und aus dem salpetersauren
Eisen entstandenes Eisenoxyd neben den anderen Beimengungen finden. Dieser Rückstand
kann dann geeignetenfalls zur Gewinnung des Silbers benutzt werden. Der Rückstand
kann nunmehr vollständig abgeröstet und schlieſslich bei der Eisenerzeugung gute
Verwendung finden. Die gewonnene Kupferlauge wird nach dem Cementationsverfahren mit
metallischem Eisen behandelt und das so erhaltene Cementkupfer weiter geläutert
(raffinirt). Die Wiedergewinnung der Salpetersäure aus den nitrosen Dämpfen bietet
nach Angabe Perino's keine Schwierigkeiten, wie weiter
unten gezeigt werden soll.
Was die Zersetzung des Eisennitrates betrifft, so ergibt der Versuch, daſs die
Reduction desselben im obigen Falle bis zur Bildung von Stickoxyd (NO) sich
erstreckt, was möglichst vollkommen eintritt, wenn die Schichtung des Materiales
eine zweckentsprechende ist. Die Umsetzung erfolgt demnach nach folgender
Reactionsgleichung:
Fe2(NO3)6 + 2 CuS = 2CuSO4 + Fe2O3 + 5NO + NO2.
Hiernach kommen theoretisch auf 1 Aeq. Cu 1 Aeq. Fe und auf 1 Th. Kupfer etwa 4 Th.
salpetersaures Eisenoxyd.
Die Zersetzung des salpetersauren Eisensalzes erfolgt ohne Beimischung eines
oxydablen Körpers erst bei Temperaturen über 100° und dann auch nur theilweise,
indem basische Salze sich bilden, welche erst bei beginnender Rothglut weiter
zerlegt werden unter Bildung nitroser Dämpfe und eines Rückstandes von Eisenoxyd.
Erleichtert wird aber die vollständige Abspaltung des Salpetersäurerestes, wenn
oxydable Substanzen zugegen sind, wie im vorliegenden Falle Schwefelmetalle. Die eigenartige und für
obigen Zweck so äuſserst vortheilhafte Wirkung des salpetersauren Eisensalzes
besteht nun weiter darin, daſs Schwefelkupfer eher und leichter durch ersteres
oxydirt wird als Schwefeleisen, Schwefelsilber u.s.w., so daſs man bei
entsprechender Durchführung des Prozesses nach dem Auslaugen eine sehr reine
Kupferlauge erhält und Eisen, Silber, Blei u.s.w., im Rückstande sich finden.
Um die Vorgänge bei der Reaction genauer verfolgen zu können, stellt man den Versuch
am einfachsten derart an, daſs man das Gemenge von Kupferkies und salpetersaurem
Eisensalze in einen Reagircylinder bringt, letzteren in die Durchbohrung der oberen
Wandung eines einfachen Trockenkastens einsenkt und die Mündung mit Kork und
Gasleitungsrohr versieht. Läſst man die Temperatur nun ganz allmählich ansteigen, so
beobachtet man bei etwas über 40° schon das Entweichen der braunrothen Dämpfe.
Erhält man die Temperatur dann etwa ½ bis 1 Stunde auf etwa 50 bis 70°, so nimmt die
Reaction ihren stetigen Fortgang, ohne daſs aber schlieſslich in dem Reagircylinder
die gefärbten Dämpfe zu sehen sind. Entfernt man aber nun den Kork, so treten sofort
an der Mündung die braunrothen Dämpfe auf, ein Zeichen, daſs sich Stickoxyd (NO)
gebildet hatte. Eine gröſsere Menge dieses Gases erhält man, wenn man zuerst die
Luft im Reagircylinder austreibt (durch Kohlensäure) und die entstehenden Gase dann
mittels des Gasleitungsrohres in einer Eudiometerröhre sammelt, Merkt man keine
weitere Einwirkung mehr, so kann man die Temperatur auf 130 bis 150° steigern, um
die letzten Reste von Nitrat zu zersetzen, was ohne Nachtheil geschehen kann, wenn
man nicht einen allzugroſsen Ueberschuſs von salpetersaurem Eisensalze zugesetzt
hatte. Letzterer bewirkt unter diesen Umständen eine Oxydation des vorhandenen
Schwefeleisens u.s.w. und entsprechende Verunreinigung der Kupferlauge. Das richtige
Mengenverhältniſs des salpetersauren Eisensalzes zum Kupferkiese ergibt sich am
sichersten durch entsprechende Vorversuche, da jenes je nach der Natur der
Kupferkiese bezieh. Kupferkies haltigen Schwefelkiese kleinen Schwankungen
unterliegen wird. Auch mag es je nach Umständen zweckmäſsig sein, den
Sulfatisirungsprozeſs in zwei Phasen zu vollziehen und danach das
Mischungsverhältniſs einzurichten, sowie die Temperaturen zu reguliren, um so eine
möglichst vollkommene Kupferextraction zu erzielen. Im Uebrigen verläuft der Prozeſs
sehr glatt und läſst sich für reiche wie arme Erze gleich gut verwerthen. Einer
besonderen Aufbereitung bedürfen die ärmeren Erze nicht und können direkt im
gemahlenen Zustande in den Prozeſs eingehen.
Treten wir nun der Darstellung des salpetersauren Eisensalzes näher, so läſst sich
dasselbe am einfachsten aus der im Cementationsprozesse erhaltenen Eisenvitriollauge
dadurch herstellen, daſs man letztere mit einer concentrirten Lösung von
salpetersaurem Kalke oder Strontian versetzt, wodurch das sehr schwer lösliche Sulfat des
Calciums bezieh. Strontiums sich ausscheidet und eine Lösung von salpetersaurem
Eisenoxydul Fe(NO3)2
sich bildet, welche in Vorlagen gebracht und durch die nitrosen Dämpfe des
Sulfatisirungsprozesses weiter zu salpetersaurem Eisenoxyd Fe2(NO3)6 oxydirt wird. Es entsprechen demnach 1Fe2(NO3)6 etwa 2Ca(NO3)2 bezieh. 2Sr(NO3)2, oder auf 1 Th. Cu kommen etwa 2½ Th.
Ca(NO3)2 bezieh.
3½ Th. Sr(NO3)2.
Zur Regeneration der Salpetersäure aus den nitrosen Dämpfen leitet man dieselben
mittels eines mäſsigen Luftstromes in mit Wasser beschickte Vorlagen, woselbst sich
verhältniſsmäſsig rasch die Bildung von Salpetersäure vollzieht, wobei man bis zu 95
Proc. zurückgewinnt, wie schon frühere Versuche von Schäppi u.s.w. dargethan haben. Diese Salpetersäure dient nun weiter zur
Herstellung des salpetersauren Kalkes bezieh. Strontians, indem man deren Carbonate
mit der Säure behandelt, wobei man unter Entwickelung von Kohlensäure die
entsprechenden Nitrate erhält, welche dann, wie weiter oben erörtert, mit der
abfallenden Eisenvitriollauge umgesetzt werden. Die frei gewordene Kohlensäure kann
zur weiteren Verwerthung dienen und ist auf diese Weise ein sehr rentables
Nebenproduct, das hierbei in groſsen Mengen erzeugt wird, indem auf 1k Kupfermetall etwa 1k Kohlensäure kommt. In der Form flüssiger Kohlensäure hat sie gegenwärtig
ein sehr bedeutendes Absatzgebiet und würde auf diese Weise eine billige Quelle zu
ihrer Herstellung verfügbar sein.
Als Abfallproduct wird demnach nur Calciumsulfat (Gyps) bezieh. Strontiansulfat
erhalten werden. Da nun wegen der besseren Fällbarkeit Strontian dem Kalke
vorzuziehen wäre, so könnte man das werthvollere Strontiumsulfat mit Sodalösung
digeriren, wodurch wieder Strontiumcarbonat und Natriumsulfat entstehen, welch
letzteres an die Sodafabriken mit Leblanc'schem
Prozesse zurückginge und den Sulfatprozeſs ersparte. Auf diese Weise wird ein
vollständiger Kreisprozeſs in der einfachsten Gestaltung erzielt und ein
Abfallproduct ganz ausgeschlossen sein. Als Rohproduct würde, bei der Verwendung von
Kalk als Fällungsmittel, der kohlensaure Kalk in seinen verschiedenen Arten ein sehr
billiges Material abgeben, wobei etwa 5 bis 8 Proc. salpetersaurer Kalk als Ersatz
für den im Prozesse entstandenen Verlust der Salpetersäure zugeschlagen werden
müſsten. Der salpetersaure Kalk wird am einfachsten in Salpeterplantagen
erzeugt.
Bei der praktischen Durchführung des neuen Verfahrens würde ein Colonnenapparat aus
Thonretorten entsprechender Form am zweckmäſsigsten erscheinen. Diese Thonretorten
befinden sich in einer gemauerten Kammer und sind durch seitliche Stutzen hinter
einander in Verbindung gesetzt, so daſs ein eingeführter schwacher Luftstrom durch
sämmtliche Retorten hindurchgeht und die nitrosen Dämpfe nach geeigneten Vorlagen
führt, in welchen sich theils salpetersaures Eisenoxydul, theils Wasser befindet. Die
durchstreichenden Gase erwärmen die Flüssigkeiten in den Vorlagen bis zu dem
gewünschten Grade, so daſs eine besondere Wärmequelle hierfür unnöthig ist. Der
Sulfatisirungsrückstand ist von sehr lockerer Beschaffenheit und läſst sich mit
groſser Leichtigkeit auslaugen.
Das vorstehend beschriebene Verfahren ist durch das D. R. P. Nr. 44498 vom 28. Januar
1888 gesetzlich geschützt worden. Der Patentanspruch lautet: „Anwendung von
salpetersauren Eisensalzen als Sulfatisirungsmittel der Kupferkiese mit oder
ohne vorherige Röstung derselben behufs Extraction des Kupfergehaltes auf nassem
Wege.“
Inwieweit dieses Verfahren für die Praxis nutzbringend sein und eine Verminderung der
Productionskosten herbeiführen wird, müssen erst im groſsen Maſsstabe ausgeführte
Versuche zeigen. Der gegenwärtige hohe Preis des Kupfers dürfte derartigen Versuchen
nicht besonders günstig sein, da bei guten Preisen die Kupferhütten meist bei den
alten, bereits bewährten Verfahren stehen bleiben, während gedrückte Preise oft die
Industriellen zwingen, auf Einführung billigerer Darstellungsmethoden zu sinnen. Der
Erfinder verspricht sich nach Angabe der Berg- und
Hüttenmännischen Zeitung neben anderen Vortheilen eine bedeutende
Erniedrigung der Gestehungskosten des Kupfermetalles und wird, wenn sein Streben von
dem praktischen Erfolge gekrönt werden sollte, gewiſs fortfahren, sein
Sulfatisirungsverfahren auch für andere Schwefelmetalle zu verwerthen.
Die Abfalllaugen von der Kupferextraction enthalten werthvolle Bestandtheile, welche
gegenwärtig meist unbenutzt bleiben.
Dr. Jurisch hat ein Verfahren zur Verarbeitung solcher
Eisenoxydul und Zink haltiger Abfalllaugen vorgeschlagen (D. R. P. Nr. 41737 vom 4.
Februar 1887).
Die Eisenoxydul und Zink haltige Lauge, welche von den Kupferfällgefäſsen abflieſst,
wird zunächst in Gefäſsen mit Rührwerk durch kohlensauren Kalk von dem gröſsten
Theile ihrer freien Säure befreit und dann in Oxydationsthürme gepumpt, welche
ähnlich eingerichtet sind wie die Weldon'schen
Apparate. Hier wird die Lauge unter gleichzeitiger Erhitzung mit Wasserdampf durch
Blasen mit Luft oxydirt, und in dem Maſse, als die Oxydation fortschreitet, indem
sich hellbraunes, basisches Eisenoxydsulfat abscheidet, wird Kalkmilch zugegeben,
ohne jedoch die Neutralisirung völlig zu erreichen. Häufig genommene Proben müssen
stets noch eine eben sichtbare Röthung auf neutralem Lackmuspapiere hervorbringen.
Durch diese Operation gelingt es, den gröſsten Theil des Eisens als unlösliches
basisches Eisenoxydsulfat niederzuschlagen. Durch Zusatz von Chlorkalk kann man die
Oxydation beschleunigen.
Das Ausfällen des Eisens als basisches Ferrisulfat kann erleichtert werden durch
Zufügung eines löslichen Sulfates, z.B. Magnesiumsulfates.
Nach Beendigung der Eisenfällung läſst man den Schlamm aus dem Oxydationsthurme in
Absetzgefäſse flieſsen, zieht die klare Zinklauge ab und schickt den Bodensatz in
eine Filterpresse.
Der Eisenniederschlag enthält als Verunreinigung eine gewisse Menge eines Zinksalzes,
weil sich an der Einlaufstelle der Kalkmilch ein momentaner Ueberschuſs von
Kalkmilch nicht vermeiden läſst – Gyps und kohlensaurer Kalk (aus der Kalkmilch) –;
man kann ihn daher zweckmäſsig an Eisengehalt anreichern, indem man ihn zum
Abstumpfen der freien Säure der nächsten Beschickung benutzt. Das auf diese Weise
gereinigte basische Eisenoxydsulfat ermöglicht die weitere Verarbeitung zu
Schwefelsäureanhydrid und Calcothar oder zu grünem Vitriole oder zu anderen
technischen Zwecken.
Die Zinklauge wird in einem Gefäſse mit Rührwerk oder Dampfstrahlgebläse heiſs mit so
viel Kalkmilch versetzt, daſs die Mischung eben deutliche Bläuung auf rothem
Lackmuspapiere hervorruft. Man läſst absetzen und pumpt den Zinkschlamm durch
Filterpressen.
Der Zinkniederschlag enthält auſser kohlensaurem Zinkoxyd noch basisches
Eisenoxydsulfat, welches der ersten Fällung entgangen ist, Mangan und kohlensauren
Kalk; ferner als in Wasser löslich: Aetzkalk, Gyps und Chlorcalcium. Er erfordert
also noch eine Aufbereitung, um dann ebenso weiter verarbeitet zu werden wie
abgeröstete Zinkblende.
Will man auch zinkarme Laugen diesem Prozesse unterwerfen, so kann man die beiden
Fällungen, nämlich des Eisens und des Zinkes, in derselben Operation bewirken und
den erhaltenen Niederschlag weiter behandeln wie den ersten Eisenniederschlag aus
zinkreichen Laugen. Die Mutterlauge vom Zinkniederschlage ist fast frei von
Schwefelsäure und enthält im Wesentlichen nur noch Chlornatrium und Chlorcalcium.
Dampft man dieselbe bis zu einer genügenden Concentration ein, so wird das
Chlornatrium unlöslich und kann ausgefischt werden; das einmal gedeckte und
getrocknete Kochsalz enthält etwa 92 Proc. Chlornatrium und kleine Mengen von
kohlensaurem Kalke, Gypse und Chlorcalcium.
Als schlieſslicher Rest dieses Prozesses bleibt (nach Angabe des Erfinders) eine
Chlorcalciumlauge, welche etwa 3108 Chlorcalcium im Liter enthält.
John Jeremiah Crooke und Robert
Crooke in New York (Nordamerika) haben ein Verfahren (D. R. P. Nr. 39920
vom 14. December 1886) angegeben, um Gold und Silber aus Kupferstein und diesen
analog zusammengesetzten Kupfererzen zu gewinnen. Dasselbe besteht im Wesentlichen
darin, daſs man die Kupfersteine oder Erze in geschmolzenem Zustande mit einer
ebenfalls geschmolzenen Bleimasse und letztere gleichzeitig mit metallischem Eisen
in Berührung hält, und zwar unter Vermeidung jeden Contactes des letzteren mit der
Kupferstein- und Erzmasse, zu dem Zwecke, um den aus letzterer übergehenden Schwefel aus dem Bleie
zu entfernen. Die fractionsweise Durchführung des Verfahrens geschieht in der Weise,
daſs man das angereicherte Blei unter Ersatz durch ärmeres Blei absticht und wieder
auf frische Kupfersteine oder Erze wirken läſst, oder daſs man die zum Theile
erschöpften Kupfersteine oder Erze unter Ersatz durch frische absticht und wieder
mit armem Bleie behandelt.
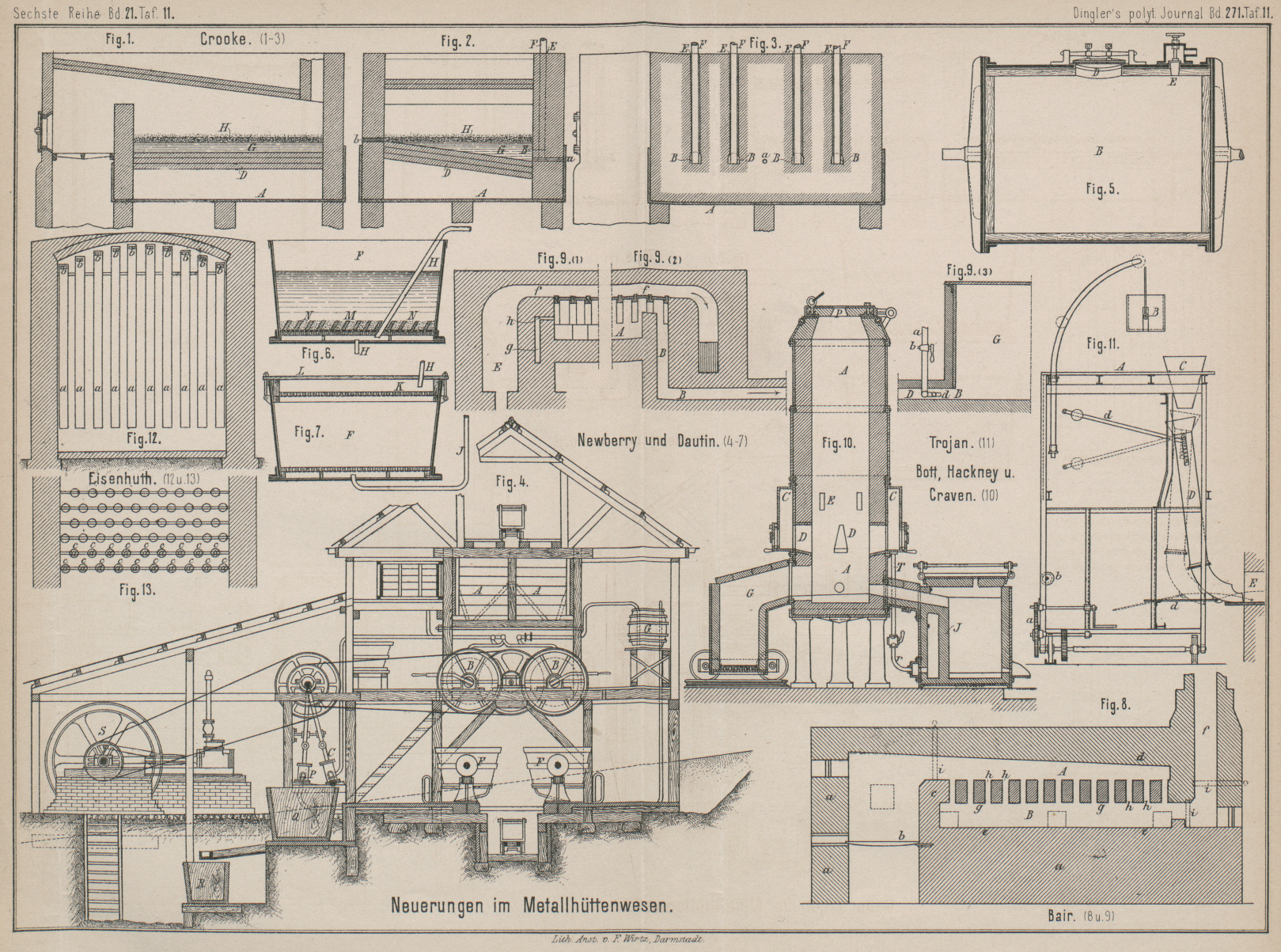
Zur Ausführung des Verfahrens dient ein in Fig. 1, 2 und 3 Taf. 11 dargestellter
Ofen; derselbe ist ein gewöhnlicher Flammofen mit fast ebenso breitem wie langem, in
eine von aufgemauerten Säulen getragene Pfanne A aus
Schmiedeeisen eingebauten Arbeitsraume mit geneigter (etwa 15cm) Sohle D. In
gleichem Niveau mit dem tieferen Ende derselben münden in den Arbeitsraum in der
betreffenden Ofenwand wagerecht angeordnete und gewölbte Kanäle B (15cm hoch und
breit bei 33cm Tiefe), deren Boden sich leicht
nach der Sohle D zu neigt. Nach oben hin setzen sich
die Kanäle B in senkrechte Kanäle E fort. Durch jeden dieser letzteren wird ein
Schmiedeeisenbarren F so eingesenkt gehalten, daſs er
auf dem Boden des betreffenden Kanales B ruht. Am
tiefen Ende der Sohle D liegt ein Abstich a für das Blei und am oberen Ende ein Abstich b für den Stein.
Zunächst wird der Arbeitsraum B mit Blei G, bis über die Kanäle B
hinaus beschickt. Nachdem dieses auf Rothglut gebracht ist, wird grob zerstückelter
Kupferstein H, in etwa dem halben Gewichte des Bleies,
auf die Oberfläche des letzteren gestreut. Der Stein geräth bald ins Schmelzen und
breitet sich dabei gleichmäſsig auf dem Bleibade aus. Will man den Kupferstein
zugleich mit der Entsilberung auch concentriren und reinigen, so wirft man 3 bis 6
Proc. Sand auf die Oberfläche des geschmolzenen Steines und knickt während einiger
Minuten leicht durch. Jetzt vollziehen sich eine Reihe von Reactionen. Das im Steine
enthaltene Gold und Silber sowie auch das Antimon und Arsen beginnen sofort sich mit
dem Bleie des Bades zu legiren. Gleichzeitig verbindet sich auch ein Theil des
Schwefelgehaltes des Steines mit Blei zu Bleisulfid, welches nun ebenso schnell, als
es sich bildet, durch das Eisen der Barren F zersetzt
wird. Das so gebildete Schwefeleisen steigt durch das Blei und den Stein an die
Oberfläche des letzteren. Ein Theil des etwa ursprünglich im Steine schon
vorhandenen Bleies oder des aus dem Entsilberungsbade in den Stein übergegangenen
Bleies verbindet sich mit der oben auf der Masse schwimmenden Kieselsäure zu
Bleisilicat, welches seinerseits das durch die theilweise Extraction des Schwefels
aus dem im Steine enthaltenen Schwefeleisen gebildete Eisensubsulfid angreift. In
dieser Weise erzeugt sich bei niederer Temperatur eine saure, sehr leicht,
schmelzbare Schlacke, die absolut kupferfrei sein soll und von Zeit zu Zeit
abgeschöpft wird. Durch dieses Mittel, sowie dadurch, daſs man in dem Maſse, wie man
Schlacke wegnimmt, wieder frischen Sand in entsprechender Menge auf wirft, wird ein groſser Theil des
im Steine enthaltenen Eisens entfernt und das Kupfer im ersteren concentrirt. Dieser
Theil des Verfahrens ist auch dann anwendbar, wenn man von den Eisenbarren F keine Anwendung macht. Während das im Bleibade aus
dem Eisen F erzeugte Schwefeleisen durch den Stein an
dessen Oberfläche steigt, wird es durch das Kupfer des Steines eines Theiles seines
Schwefels wieder beraubt, an der Oberfläche sodann von dem Bleisilicate angegriffen
und in die Schlacke übergeführt. Die Erfinder lassen die vorstehend geschilderten
Reactionen etwa 80 Minuten lang vor sich gehen. Während dieser Zeit sollen aus dem
Steine etwa 90 Proc. Silber und Gold und, falls er Blei enthielt, auch der gröſste
Theil von diesem ausgezogen werden. Um einige Zahlenbeispiele zu geben, möge
folgendes angeführt werden.
Enthielt der Stein z.B. 6220g Silber in der Tonne
und 30 Proc. Blei, so gingen während der genannten 30 Minuten in das Extractionsbad
etwa 5440g Silber und etwa 168 bis 186k kupferfreies aber mit dem gröſsten Theile des im
Steine vorhandenen Antimons und Arsens beladenes Blei über. Wurden auf 1t Stein 2t Blei
angewendet, so beträgt der Verbrauch an Eisen aus dem Barren F etwa 34 bis 37k. Wurden aus dem Steine
186k Blei reducirt, so wurden auf 37k reducirtes Blei 9 bis 10k Eisen aus den Enden der Barren F verbraucht.
Wird beim Beginne der Arbeit zur Extraction reines, d.h. von Antimon, Arsen u.s.w.
freies Blei verwendet, so kann man durch eine einmalige Anwendung dieses Bleies
praktisch den ganzen Silber- und Goldgehalt des Steines extrahiren. Um jedoch die
Kosten für das Raffiniren so groſser Bleimassen zu vermeiden, ziehen die Erfinder es
vor, für die erste Behandlung von frischem Steine Blei anzuwenden, welches bereits
zu einer vorhergehenden Operation benutzt worden ist und dementsprechend mehr oder
weniger Antimon, Arsen und auch etwas Kupfer enthält. Dasselbe soll schon dem Steine
etwa 90 Proc. der Edelmetalle entziehen; die übrigen 10 Proc. werden dann durch
frisches und darum energischer wirkendes Blei ausgezogen.
Die Eisenbarren hebt man, nachdem das bereits benutzte Blei auf den frischen Stein
etwa 30 Minuten lang eingewirkt hat, heraus und sticht dieses Blei durch a ab. Dasselbe wird nochmals zur ersten Behandlung von
frischem Steine benutzt oder raffinirt. Dann beschickt man den Ofen mit dem gleichen
Gewichte noch nicht zur Extraction benutzten Bleies, senkt die Barren F wieder ein und läſst 10 Minuten wirken, nachdem man
einen Augenblick durchgekrückt hat. Der jetzt entsilberte und concentrirte Stein
wird in b abgestochen und, wie üblich, in Sandformen
laufen gelassen. Die Bleibeschickung beläſst man dagegen im Ofen. Man besetzt sie
mit frischem, noch nicht behandelten Steine und wiederholt den Prozeſs. Entsilberter
und concentrirter Stein wird nach bekannten Verfahren zu Gute gemacht.
Dr. Emil Wohlwill, langjähriger Leiter der
elektrolytischen Anstalt der Norddeutschen Affinerie in Hamburg, hat in der am 20.
Juni 1888 abgehaltenen Sitzung des Naturwissenschaftlichen
Vereines zu Hamburg über das Zerfallen der Anode bei der Elektrolyse etwa
Nachstehendes mitgetheilt.
Wird bei der Zersetzung von verdünnter Schwefelsäure oder einer Lösung von
Kupfervitriol durch den elektrischen Strom die Anode aus reinem Kupfer genommen, so
bedeckt sich dieselbe, so lange die elektrochemische Auflösung dauert, mit einem aus
feinvertheiltem metallischem Kupfer bestehenden rothen Staube, der allmählich
abfällt. Da gleichzeitig die Anode, je länger die Arbeit dauert, um so mehr gefurcht
und zerfressen wird, so scheint der Vorgang, der in ähnlicher Weise bei anderen
reinen Metallen beobachtet wird, sich den zuerst von Delarive (1837) beschriebenen, aber bisher nicht genügend erklärten
Erscheinungen der Desaggregation der Elektroden anzuschlieſsen. Als nothwendige
Folge des besprochenen Verhaltens ist anzusehen, daſs im scheinbaren Widerspruche
mit dem Faraday'schen Gesetze, insbesondere bei der
Elektrolyse von Kupferlösungen die Gewichtsabnahme der Anode gröſser ist, als die
Zunahme der Kathode. Da ein regelmäſsiges Abfallen ungelöster Theile bei der
gewöhnlichen chemischen Lösung reiner Metalle nicht stattfindet, ist die Erklärung
für das Verhalten der Kupferanode in den Besonderheiten der elektrochemischen
Auflösung zu suchen; als solche sind zu betrachten, daſs bei der elektrolytischen
Auflösung das Lösungsmittel stets im „Entstehungszustande“ wirkt, daſs es nie
im Ueberschusse vorhanden ist und daſs es sich für Verschiedenheiten des chemischen
und elektrischen Verhaltens der Anodenbestandtheile äuſserst empfindlich zeigt.
Darauf läſst sich zur Erklärung der besprochenen Erscheinung die folgende Annahme
gründen: Wie thatsächlich eine heiſse Lösung von Kupfersulfat in Berührung mit
metallischem Kupfer Theile desselben aufnimmt, Oxydulsulfat bildet, das beim
Abkühlen wieder in Kupfer und Kupfervitriol (Cuprisulfat) zerfällt, so wird auch
unter dem Einflüsse des Entstehungszustandes an der Anode neben Kupfervitriol die
metallreichere Verbindung gebildet, die dann auſserhalb der Entstehungssphäre unter
Abscheidung eines fein vertheilten Kupferniederschlages wieder zerfällt. Diese
Erscheinung wäre demnach nicht auf Desaggregation der Anode selbst, sondern auf
Dissociation der an ihr gebildeten Lösung zurückzuführen. Mit dieser Erklärung sind
die quantitativen Verhältnisse des Anodenfalles im Einklänge. Die abfallende Menge,
annähernd durch den Mehrverlust der Anode zu messen, ist um so gröſser, je gröſser
der Säuregehalt der Lösung; das Gleiche gilt für die Wirkung einer heiſsen
Kupfervitriollösung auf metallisches Kupfer. Der Ueberschuſs des Anoden Verlustes
ist ferner um so gröſser, je kleiner die Stromdichte. Dies entspricht der
Vorstellung, daſs, wie in vielen analogen Fällen thatsächlich nachgewiesen wurde,
auch hier die Menge der
an der Anode entstehenden metallreichen Verbindung neben der metallärmeren bei
wachsender Stromdichte sich verringert. Es ist endlich der Mehrverlust und der
Abfall der Anode niemals der Zeitdauer der elektrochemischen Wirkung proportional,
sondern um so kleiner, je länger die Wirkung auf die unberührte Anode dauert. Dies
erklärt sich durch die Voraussetzung, daſs eine Erhaltung ungelöster Metalltheile an
der Oberfläche der Anode überhaupt nur möglich ist, wenn dieselben gegen das in
Lösung übergehende Metall der Anode sich elektronegativ verhalten. Bedeckt sich in
Folge dessen die Oberfläche der Anode mehr und mehr mit gewissermaſsen weniger
löslichen Theilen, so wird nach weniger Zeit die freiliegende positivere Oberfläche
nicht mehr der Menge des elektrisch abgeschiedenen Lösungsmittels genügen und dann
auch der negativere Metallstaub mitgelöst, also die Menge des Anodenabfalles
verkleinert werden. Der gegebenen Erklärung gemäſs müſsten weder ein Zerfallen der
Anode, noch Schwankungen des Gewichtsverlustes der bezeichneten Art zu beobachten
sein, wenn die an der Anode entstehende metallreichere Verbindung auch auſserhalb
ihrer Entstehungssphäre beständig ist. Diesen Erwartungen entspricht das Verhalten
einer Kupferanode, wenn an derselben als Zersetzungsproduct der Salzsäure Chlor
abgeschieden und Kupferchlorür gebildet wird (Berg- und
Hüttenmännische Zeitung, 1888 S. 257).
Paul Mellmann in Berlin hat ein Verfahren zur Darstellung von Phosphorkupfer oder Phosphorzinn (D. R. P. Nr. 45175 vom 8. Februar 1888) vorgeschlagen. Dasselbe besteht im Wesentlichen darin, daſs in einer Operation
phosphorsaure Alkalien mittels Kieselsäure und Kohle in kieselsaure Alkalien und
freien Phosphor verwandelt werden und dieser im Entstehungszustande an die erwähnten
Metalle gebunden wird. Folgende Gleichung gibt den einzuschlagenden Weg an:
x(2ROP2O5 + SiO2 + 10C) +
yMe = xR2O2SiO2 + x2PyMe + x10CO.
Nach dem sorgfältigen Zerkleinern der Phosphorsäure haltigen Materialien (Apatit,
Phosphorit, Knochen oder auch Thomasschlacke) werden dieselben nach vorstehender
Formel gemischt – wobei auch statt des Metalles selbst eine Sauerstoffverbindung
desselben benutzt werden kann, was für den Zusatz der Kohle in Berechnung zu ziehen
ist – und auf den Herd eines Flammofens oder in einen Tiegel gebracht und
eingeschmolzen. Das Phosphormetall sammelt sich dann unter den Silicaten an und kann
abgestochen werden. Um Ersparnisse an Brennmaterial zu machen, ist es zweckmäſsig,
ein Fluſsmittel (Soda oder Fluſsspath) zuzugeben. Dasselbe hat keinen anderen Zweck,
als die Zersetzung einzuleiten.
Soll die Gewinnung im Gebläseschachtofen vorgenommen werden, so wird an Stelle des
Sandes ein fetter Lehm genommen. Mittels desselben wird die Masse gebunden und
geformt, wodurch einerseits ein Fortblasen der staubförmigen Gemengetheile
vermieden, andererseits ein leichter Ofengang erzielt wird. Das Gemenge wird, wie gewöhnlich, mit Koks
geschichtet in die Oefen eingetragen, auf seinen Schmelzpunkt erhitzt und sickert
dann als Schlacke bezieh. Metall durch das Brennmaterial bis auf die Sohle des
Ofens, woselbst die Scheidung in Schlacke und Phosphormetall vor sich geht und von
wo aus dieselben abgestochen werden.
Gold und Silber. Um schwer aufschlieſsbare Gold und
Silber führende geschwefelte Erze auf ihren Edelmetallgehalt zu Gute zu machen, wird
nach dem Vorschlage von Alexander Parkes in Dulwich,
England (D. R. P. Nr. 45013 vom 15. September 1887) das Erz nur gröblich zerkleinert
und dann in einem Flammofen niedergeschmolzen unter Zuschlag von kaustischen Erden
und kaustischen Alkalien (bezieh. Alkali-Carbonat, -Nitrat, -Sulfat oder -Silicat)
und Kohle. Das hierbei sich ergebende Zwischenproduct wird dann nach bekannter
Methode mit metallischem Blei oder mit Bleisauerstoffverbindungen oder
Bleischwefelverbindungen oder anderen für diesen Zweck bekannten Metallen oder
Metall Verbindungen weiter behandelt.
Das nach obiger Methode erzeugte Zwischenproduct soll die charakteristische
Eigenschaft besitzen, zu einem feinen Pulver zu zerfallen, wenn man es einige
Stunden der Luft aussetzt. Durch Besprengen des noch heiſsen Zwischenproductes mit
Wasser wird das Zerfallen noch gefördert. In diesem Zustande ist dann das
Zwischenproduct für die Behandlung mit Blei sehr geeignet.
Das zweckmäſsige Verhältniſs des Zuschlages zum Erze ergibt sich aus der Fassung des
Patentanspruches, welcher folgendermaſsen lautet:
„Die Erzeugung eines an der Luft zerfallenden Zwischenproductes durch
Einschmelzen eines nicht abgerösteten, geschwefelten, Gold oder Silber führenden
Erzes mit 12 bis 16 Proc. kaustischem oder kohlensaurem Kalke und 5 Proc.
kaustischem Natron.“
Anstatt eines Flammofens kann man das Erz auch in einem Cupol- oder Gebläseofen
niederschmelzen. Das Wesentliche besteht nur immer darin, daſs die Erze nicht
abgeröstet zu werden brauchen. Das vorstehend beschriebene Verfahren stellt sich
also als eine Abänderung der bekannten Goldgewinnung durch Schmelzprozeſs dar, bei
welcher Methode in jüngster Zeit wenig Neuerungen aufgetreten sind, da das Interesse
sich selbstverständlich vorwiegend den nassen Prozessen zuwendet.
In D. p. J. 1888 269 368, 578
ist bereits von dem Verfahren zur Golgewinnung mittels Chlores von Newberry und Vautin
berichtet worden. Fig. 4 veranschaulicht die zu dem Verfahren erforderlichen Einrichtungen
in ihrer Gesammtheit, und Fig. 5, 6 und 7 Taf. 11 die Theile
derselben nach dem D. R. P. Nr. 44913, gültig vom 7. Januar 1888.
Das kleinkörnige, Gold haltige Material wird mittels Rührwerkes A in die Extrationskessel B (Fig.
4 und 6) geleitet, welchen zur Förderung des Extractionsprozesses eine langsame
Drehung ertheilt wird. Die eisernen Wandungen dieser Kessel sind auf der inneren
Seite, wie bereits früher mitgetheilt, zum Schütze gegen die Einwirkung des Chlores
mit Blei bekleidet. Es empfiehlt sich, das Bleihemd mit einem Futter aus Holz oder
Steingut zu bedecken, um die Beschädigung desselben durch die in Bewegung gehaltene
Füllung der Kessel zu verhüten. Zur Einführung des zu behandelnden Materiales dient
ein Mannloch D und zur Einleitung des Chlorgases und
der gepreſsten Luft das Sperrventil E. Zur Förderung
der Luft wird eine Compressionspumpe C angewendet.
Ist der Extractionsprozeſs vollendet, so wird das in den Kesseln B noch enthaltene Gemenge von Gas und Luft nach einem
mit Kalkwasser gefüllten Bottiche G geleitet, um die
Ansammlung schädlicher Gase im Arbeitsraume zu verhüten. Nachdem dies geschehen,
werden die Kessel B nach Oeffnung der Mannlöcher in die
Filterbottiche F (Fig. 4, 5 und 6) entleert. Hier erfolgt
die Scheidung der flüssigen Goldlösung von den erdigen Substanzen, und zwar indem
die Filtrirung entweder in der Richtung nach unten oder in der Richtung nach oben
geschieht. Fig.
6 und 7 zeigen die Gestalt des Filters in ersterem bezieh. in letzterem Falle.
In beiden Fällen besteht das Filterbett M aus einem
rostartig durchbrochenen Holzboden, welcher mit Canevas oder einem Asbestgewebe
bedeckt ist. Der Filterbottich nach Fig. 6 kann offen bleiben,
derselbe nach Fig.
7 (Filtrirung nach oben) wird mit einem luftdicht schlieſsenden Deckel
versehen; ersterer enthält das Filterbett M in seinem
unteren Theile, letzterer in seinem oberen Theile bei K. Zur Beschleunigung der Filtrirarbeit wird die Flüssigkeit unter
Anwendung einer Saugepumpe durch das Filter gesaugt. Das Saugerohr H mündet bei Fig. 6 in den Raum
zwischen Filterbett und Bottichboden und bei Fig. 7 in den Raum
zwischen Filterbett und Bottichdeckel; bei Fig. 6 sind zwei
Saugerohre H angegeben, die abwechselnd in Betrieb
genommen werden können.
Um die Nutzwirkung des Filters zu erhöhen, wird die Füllung der Filterbottiche durch
Wasser verdünnt. Geschieht die Filtrirung in der Richtung nach oben (Fig. 7), so findet die
Einführung des Wassers mittels eines Rohres J statt,
welches in den an seinem oberen Theile mit Aussparungen versehenen Doppelboden des
Filterbottiches einmündet.
Ist die Filtrirung vollendet, so erfolgt die Entfernung des Rückstandes der Filter
durch Umwenden derselben. Damit hierbei das Filterbett nicht in Unordnung geräth,
sind über demselben schräg gestellte Holzleisten M
(Fig. 6)
angebracht, welche die Canevasbedeckung in ihrer richtigen Lage erhalten.
Die filtrirte Goldflüssigkeit wird in eine Bütte Q
geleitet, in welche ein Strahl Dampf oder Luft eingeführt wird, um freies Chlor,
welches in der Flüssigkeit noch enthalten sein könnte, abzutreiben.
Die Flüssigkeit gelangt endlich in die bei B
angedeuteten Kohlenfilter, wo, wie beschrieben, die Ausscheidung des metallischen
Goldes und dessen Niederschlagung auf die Holzkohlenfüllung erfolgt.
Der Deutschen Gold- und Silber-Scheideanstalt vormals
Roessler in Frankfurt a. M. ist unter Nr. 45194 ein
D. R. P. vom 3. Mai 1888 auf ein Verfahren zum Feinmachen von Blicksilber mit
folgendem Patentansprüche verliehen worden: „Das Feinmachen von Blicksilber durch
Einrühren von schwefelsaurem Silberoxyd und die Abscheidung bezieh. Gewinnung
von in dem Blicksilber enthaltenem Blei und Wismuth in Form von Sulfat.“
Das Blicksilber wird in Graphittiegeln eingeschmolzen und in die Oberfläche des
Metallbades allmählich geschmolzenes schwefelsaures Silber eingerührt. Es tauschen
sich dann Blei und Wismuth in dem Sulfate aus und gehen in die obenauf schwimmende
Schlacke.
Um das Angreifen des Graphittiegels zu verhüten, gibt man vorher auf das geschmolzene
Metall eine Schicht von Quarzsand, in deren Mitte man das Silbersulfat einrührt, und
welches das gebildete Blei- und Wismuthsulfat aufnimmt und damit eine zähe,
dickflüssige Schlacke bildet, die wiederholt abgehohen wird, bis das Silber ganz
fein ist.
Im Anfange geht wesentlich nur Blei in die Schlacke, etwa vorhandenes Wismuth aber
erst am Ende des Prozesses. Man kann deshalb die letzten Schlacken, welche das
Wismuth angereichert enthalten, gesondert aufbewahren und dasselbe leicht daraus
gewinnen.
Das schwefelsaure Silber stellt man in einem eisernen Kesselchen dar durch Auflösen
von Feinsilber in Schwefelsäure, Verdampfen der überschüssigen Säure und Erhitzen
der Masse bis zum Schmelzen. Es wird dann ausgegossen und nach dem Erkalten in
Stücke geschlagen.
An dieser Stelle mögen noch einige kleinere Mittheilungen Platz finden. E. Matthey trennt Gold und Silber dadurch von Wismuth,
daſs er 2 Proc. Zink zu dem geschmolzenen Metalle hinzufügt, die Masse allmählich
abkühlen läſst und die Oberfläche reinigt. Dieses Verfahren wird wiederholt. Das
ganze Gold und Silber geht in den Schaum. Die so erhaltene Wismuthglätte wird in
einem Tiegel mit Borax geschmolzen. Hierbei sinkt das Gold auf den Grund, indem es
gleichzeitig von irgend welchen unedlen Metallen durch die Gegenwart des
Wismuthoxydes befreit wird. Die Schlacke schmilzt man wieder mit Wismuth zusammen,
um die letzten Spuren von Gold zu entfernen.
Wismuth wird vom Bleie mittels wiederholten Auskrystallisirens getrennt, da
Legirungen von Wismuth und Blei bei niedrigerer Temperatur schmelzen als Wismuth
selbst. Ein Wismuth, welches 4 Proc. Blei enthielt, besaſs nach vier
Krystallisirungen nur 0,4 Proc. (Engineering and Mining
Journal, 1888 S. 349).
Edward Matthey hat in einer Sitzung der Royal Society darauf hingewiesen, daſs metallisches
Wismuth häufig eine kleine Menge von Kupfer enthält, welches die guten Eigenschaften
dieses Metalles sehr beeinträchtigt.
Da die Entfernung dieser geringen Mengen von Kupfer auf nassem Wege zu langwierig und
kostspielig ist, so wendet Matthey ein schnelles und
einfaches trockenes Verfahren an, um ganz reines Wismuth zu erlangen. Von Arsen,
Antimon, Blei und Tellur wird das Wismuth zuerst auf bekannte Weise gereinigt, dann
schmilzt er es mit Schwefelwismuth zusammen. Der Schwefel verbindet sich sofort mit
dem Kupfer. Man soll auf diese Weise ein ganz reines Wismuth erlangen können aus
einem Materiale, welches 0,1 bis 1 Proc. Kupfer enthält (Industries vom 3. Februar 1888).
Zur Trennung des Zinnes vom Antimon und Bestimmung desselben in siliciumhaltigen
Schlacken und Legirungen werden nach H. N. Warren (Chemical News, 1888 Bd. 57 S. 124) 2g der fein gepulverten Schlacke in einer
Platinschale mit einem Gemische gleicher Theile Fluſssäure und Salzsäure behandelt,
wobei die Schlacke in wenigen Minuten zersetzt und der gröſsere Theil der
Kieselsäure als SiF4 verflüchtigt wird, während der
Rest und ebenso alles Zinn in Lösung geht. Man filtrirt, erwärmt, sättigt mit H2S, kocht den Niederschlag behufs Abscheidung von
Wismuth und Kupfersulfid mit Natron und fällt die Lösung mittels Salzsäure. Die
abgeschiedenen Sulfide von Antimon und Zinn werden jetzt mittels Königswassers in
Lösung genommen, worauf man behufs Vertreibung überschüssiger Salpetersäure auf ein
kleines Volumen eindampft, mit mäſsig schwacher wässeriger Salzsäure versetzt, einen
Ueberschuſs von Kaliumferrocyanid zufügt und die Lösung, welche, wenn genügend K4FeCy6 zugesetzt
ist, rein blau erscheint, kocht. Hierbei wird alles Zinn als Stanniferrocyanid
gefällt, während das Antimon in Lösung bleibt und durch Schwefelwasserstoff gefällt
werden kann. Der Zinnniederschlag wird getrocknet und geglüht, wobei man zur
schnellen Zerstörung der organischen Substanz einige Tropfen Salpetersäure zufügt.
Den Rückstand reducirt man im Tiegel mit tubulirtem Deckel durch Wasserstoff oder
Kohlengas, läſst erkalten, löst in Salzsäure, fällt das Zinn als Sulfid, oxydirt mit
Salpetersäure und bestimmt es in üblicher Weise. Die Trennung von Zinn und Antimon
in Legirungen kann in derselben Weise vorgenommen werden, nur löst man die Legirung
in Königswasser, statt in Salz- und Fluſssäure. Das nach der Abscheidung des Zinnes
mittels K4FeCy6
gefällte Schwefelantimon muſs hell orangeroth, nicht aber bräunlich gefärbt sein
(Oesterreichische Zeitschrift für Berg- und
Hüttenwesen, 1888 S. 512).
(Schluſs folgt.)
Tafeln